Session Information
Session Type: Poster Session B
Session Time: 9:00AM-11:00AM
Background/Purpose: The mucosal microbiome contributes to disease pathogenesis via local and systemic interaction with the host. The hallmark of this interaction in the physiological condition is the varying ability of bacterial products to translocate into the circulation via mucosa. Thus, the blood microbiome is important to investigate microbial-host interactions and the effects on systemic immune perturbations. However, this effort has met with major challenges due to low microbial biomass and background artifacts.
Methods: In the current study, microbial 16S DNA sequencing was applied to analyze the plasma microbiome. We have developed a quality-filtering strategy to evaluate and exclude low levels of microbial sequences, potential contaminations, and artifacts from plasma microbial 16S DNA sequencing analyses. Furthermore, we have applied our technique in individuals with systemic lupus erythematosus (SLE) and corresponding healthy controls.
Results: We first analyzed the potential contamination sources from the collection of blood samples, microbial DNA extraction, to microbial DNA sequencing. Notably, the β-diversity of plasma microbial 16S rDNA was significantly different from those of the controls (P < 0.001, Multivariate Welch t-test, Figure 1). To identify the microbial amplicon sequence variants (ASVs) that are associated with disease pathogenesis, 97%-99% of total sequence data was removed using stringent quality-filtering strategy analyses; those removed ASVs were low levels of microbial sequences, contaminations, and artifacts (Figure 2). The specifically enriched pathobiont bacterial ASVs have been identified in plasmas from individuals with SLE but not from the control subjects (Nonparametric Mann-Whitney’s U tests, Figure 3). The associations between these ASVs and plasma levels of SLE-related autoantibodies were demonstrated (Spearman correlation tests, P < 0.05, Figure 3).
Conclusion: The whole-genome sequencing technique has been applied to analyze the blood microbiome (Poore GD, et al, Nature, 2020); in this study, more than 90% of total sequence data was removed after decontamination analysis. In the current study, we present a quality-filtering strategy to identify abundant plasma microbial ASVs. This strategy can be used to discover translocated microbial DNAs that correlate with biological conditions. Our approach provides a cost-saving method for the diagnosis of subclinical microbial infection as well as for understanding the roles of microbiome-host interaction in disease pathogenesis.
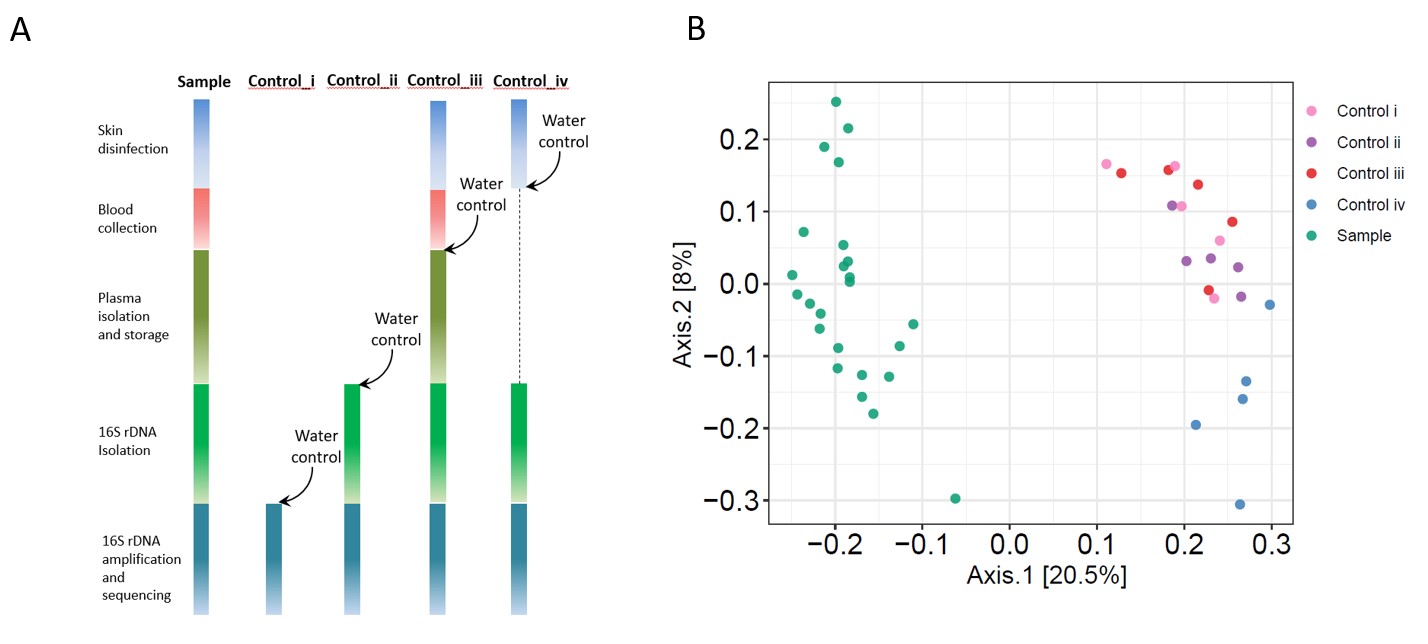 Figure 1. Overall 16S rDNA plasma microbiome analysis. (A) A workflow diagram is shown to collect blood samples and each control. (B) Principal coordinate analysis (PCoA) was conducted based on the unweighted UniFrac distance to determine the beta diversity of plasma microbial community among each controls and plasma samples.
Figure 1. Overall 16S rDNA plasma microbiome analysis. (A) A workflow diagram is shown to collect blood samples and each control. (B) Principal coordinate analysis (PCoA) was conducted based on the unweighted UniFrac distance to determine the beta diversity of plasma microbial community among each controls and plasma samples.
 Figure 2. Strategies for removing background and potential artifacts from plasma microbiome. (A) A workflow diagram of user-defined quality-filtering strategy to exclude the background and artifacts. (B) The number of removed amplicon sequence variants ASVs with different filtering factors, we chose the filtering factor of 6 which showed the lowest number of removed ASVs during the plateau effect. (C) The abundance and prevalence of ASVs in each step of filtration. The “exclude 1” step is removing low abundance of ASVs across samples, in which a 0.005% minimum abundance threshold was applied. The “exclude 2” step is removing low prevalence of ASVs in each sample; we retained ASVs only if its prevalence was more than 1/3 across study groups. If the prevalence of ASVs was lower than 1/3 in both control and case groups, it would be removed, because it was not considered the representative disease associated ASVs. The “exclude 3” step is removing potential contaminants and artifacts using filtering factors.
Figure 2. Strategies for removing background and potential artifacts from plasma microbiome. (A) A workflow diagram of user-defined quality-filtering strategy to exclude the background and artifacts. (B) The number of removed amplicon sequence variants ASVs with different filtering factors, we chose the filtering factor of 6 which showed the lowest number of removed ASVs during the plateau effect. (C) The abundance and prevalence of ASVs in each step of filtration. The “exclude 1” step is removing low abundance of ASVs across samples, in which a 0.005% minimum abundance threshold was applied. The “exclude 2” step is removing low prevalence of ASVs in each sample; we retained ASVs only if its prevalence was more than 1/3 across study groups. If the prevalence of ASVs was lower than 1/3 in both control and case groups, it would be removed, because it was not considered the representative disease associated ASVs. The “exclude 3” step is removing potential contaminants and artifacts using filtering factors.
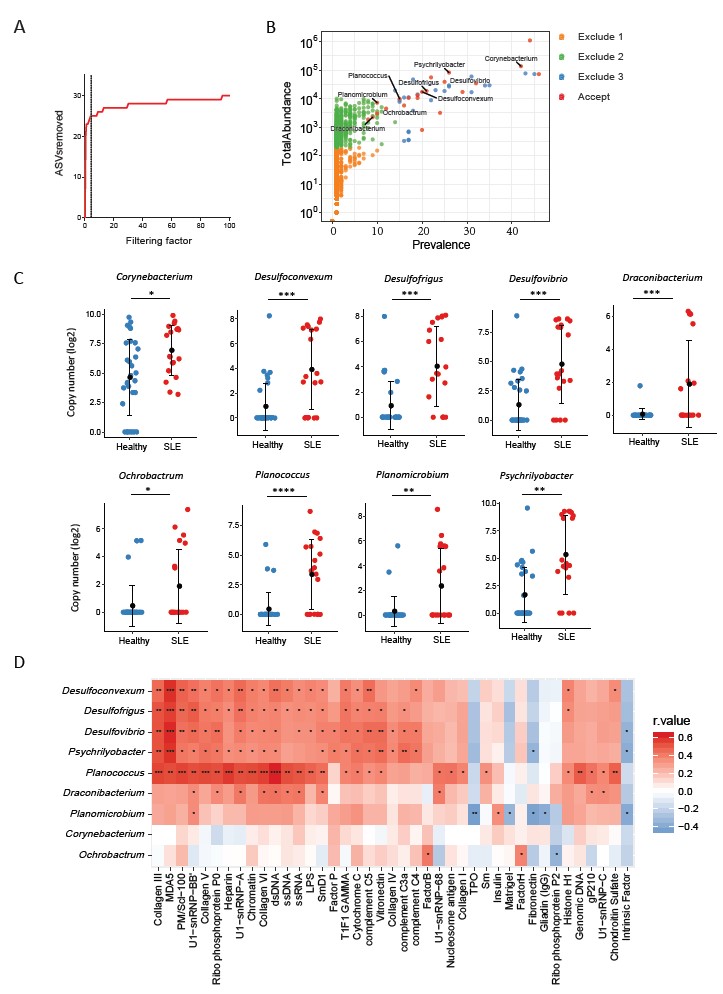 Figure 3. Plasma microbiome in female SLE patients and healthy female controls. (A) The number of removed ASVs with different filtering factor, we chose the filtering factor of 3 which showed the lowest number of removed ASVs during the plateau effect. (B) The abundance and prevalence of ASVs in each step of plasma microbiome filtration are shown, and the significantly different taxa in SLE patients compared with healthy controls are labeled in black (enriched in SLE patients). (C) The significantly different taxa in SLE patients compared with healthy controls. (D) Correlations between plasma levels of autoantibodies and differential plasma taxa levels in the two study groups. Correlation coefficient r values are indicated by color from red (directly correlations) to blue (inversely correlations); p-value significance is shown in the heatmap. Nonparametric Mann-Whitney’s U and Spearman’s rank tests. * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001.
Figure 3. Plasma microbiome in female SLE patients and healthy female controls. (A) The number of removed ASVs with different filtering factor, we chose the filtering factor of 3 which showed the lowest number of removed ASVs during the plateau effect. (B) The abundance and prevalence of ASVs in each step of plasma microbiome filtration are shown, and the significantly different taxa in SLE patients compared with healthy controls are labeled in black (enriched in SLE patients). (C) The significantly different taxa in SLE patients compared with healthy controls. (D) Correlations between plasma levels of autoantibodies and differential plasma taxa levels in the two study groups. Correlation coefficient r values are indicated by color from red (directly correlations) to blue (inversely correlations); p-value significance is shown in the heatmap. Nonparametric Mann-Whitney’s U and Spearman’s rank tests. * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001.
To cite this abstract in AMA style:
Jiang W, Alekseyenko A, Gilkeson G, Oates J, Ogunrind E, Li Q, Kamen D, Tsao B, Luo Z. Rigorous Plasma Microbiome Analysis Method Enables Disease Association Discovery [abstract]. Arthritis Rheumatol. 2020; 72 (suppl 10). https://acrabstracts.org/abstract/rigorous-plasma-microbiome-analysis-method-enables-disease-association-discovery/. Accessed .« Back to ACR Convergence 2020
ACR Meeting Abstracts - https://acrabstracts.org/abstract/rigorous-plasma-microbiome-analysis-method-enables-disease-association-discovery/