Session Information
Date: Monday, November 13, 2023
Title: (1221–1255) Pediatric Rheumatology – Clinical Poster II: Connective Tissue Disease
Session Type: Poster Session B
Session Time: 9:00AM-11:00AM
Background/Purpose: JDM is a rare immune-mediated disease of childhood that is thought to result from genetic predisposition and environmental drivers, with documented links to microbial exposures. In this multi-center, prospective, observational cohort study, we evaluated whether JDM is associated with discrete oral and gut microbiome signatures using siblings to control for family-associated microbial communities.
Methods: We generated 16S rRNA sequence data from fecal, saliva, supragingival, and subgingival plaque samples from JDM probands (n=28, age range 3-18 years, mean age 10 years, 46% female). To control for genetic and environmental determinants of microbiome community structure, we also profiled microbiomes of their unaffected family members (n=27 siblings, n=26 mothers, and n=17 fathers). We performed paired within-family comparisons as well as unpaired analyses of different cohorts.
Results: Family unit was the predominant factor explaining variance in both weighted and unweighted UniFrac distances between samples for each sample type (p< 0.001 in fecal, saliva, supragingival swab, and posterior subgingival dental plaque samples; p< 0.01 in anterior subgingival dental plaque samples, permutational multivariate analysis of variance. Unweighted UniFrac distances between siblings was significantly smaller than the distances between JDM probands in our cohort (p< 0.002, two-tailed Student’s t-test).
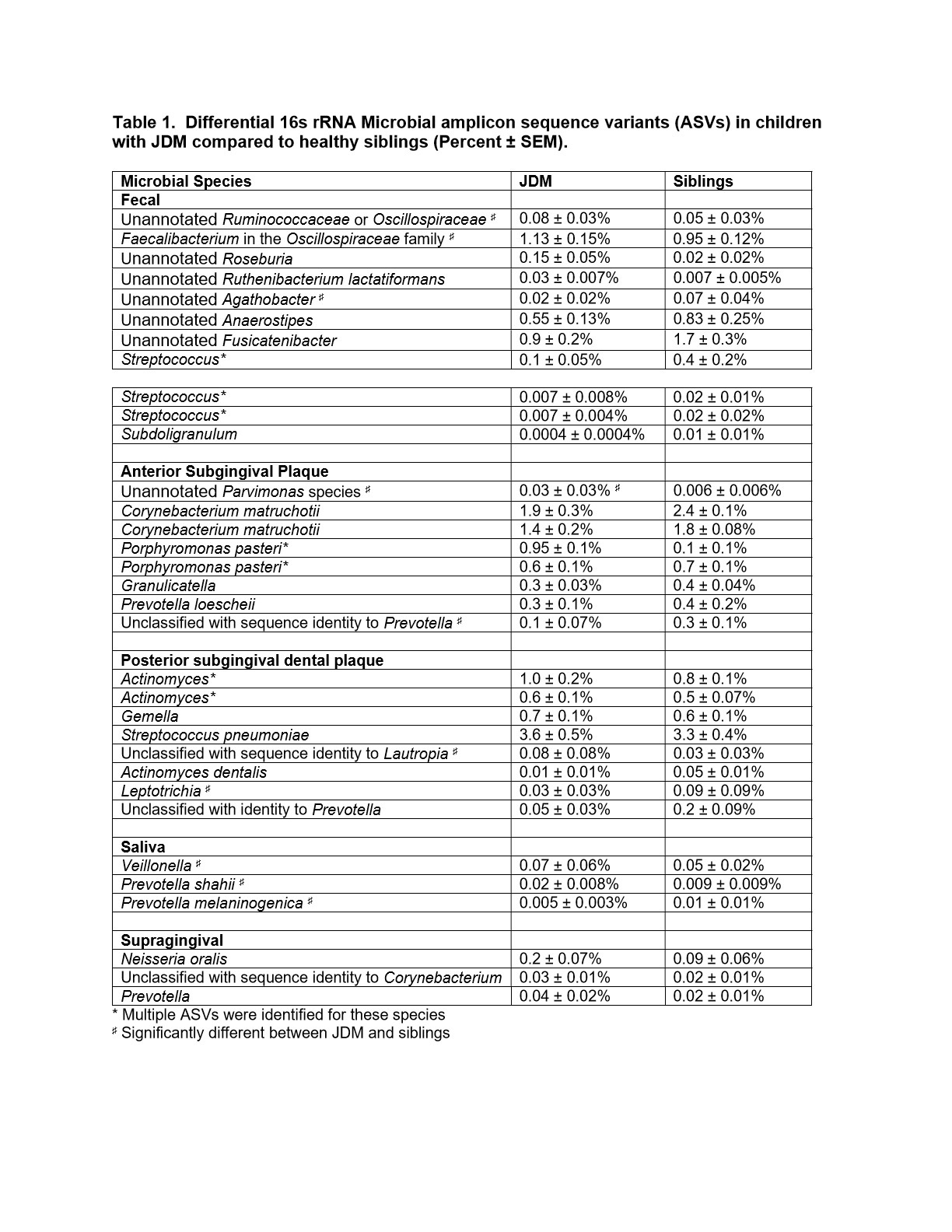
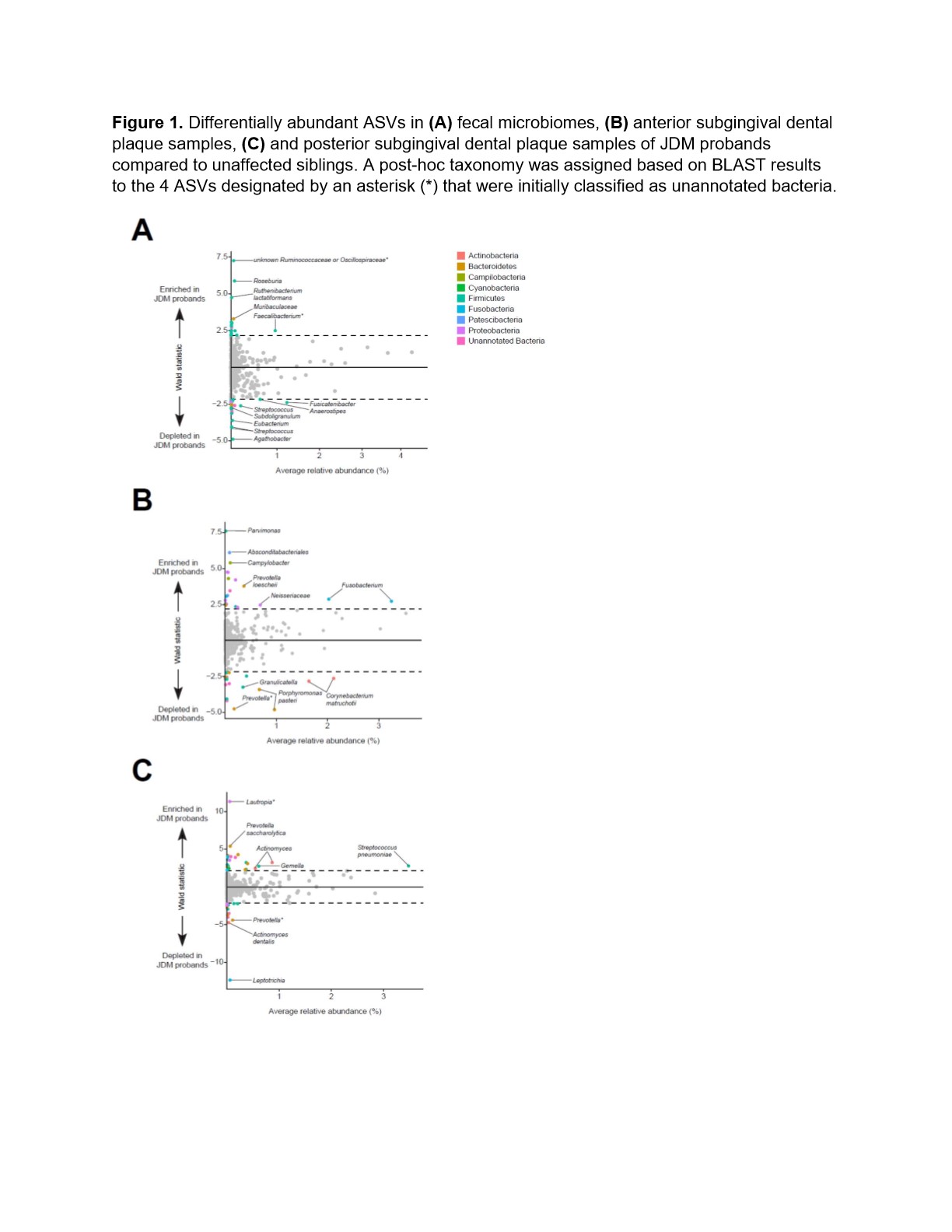
In fecal samples, 4% of bacterial amplicon sequence variants (ASVs) were differentially abundant in microbiomes between JDM probands and their siblings (Figure 1A, Table 1). In the case where ASVs that were initially classified as unannotated bacteria during the analyses, taxonomy was assigned based on BLAST results.Approximately 8% of ASVs identified in the anterior subgingival dental plaque were significantly different between JDM probands and their healthy siblings (Figure 1B). In posterior subgingival dental plaque samples, approximately 7% of identified ASVs were significantly differentially abundant between JDM probands and their siblings (Figure 1C). We also observed differentially abundant ASVs in the saliva and supragingival swab samples from JDM probands, all of which were found in very low abundances, including salivary enrichment of Veillonella and Prevotella shahii and depletion of Prevotellamelaninogenica.
Conclusion: The oral and gut microbiomes of JDM probands were more similar to their own unaffected siblings than they were to the microbiomes of other JDM probands, suggesting family unit has a significant effect on microbiome community structure. Nonetheless, in a sibling-paired analysis, several potentially immunomodulatory bacterial taxa were differentially abundant in the microbiomes of JDM probands, including Faecalibacterium in the gut and Streptococcus in the oral cavity. The loss or gain of specific fecal and oral bacteria may potentially play a role in disease pathogenesis and/or be secondary to immune dysfunction in susceptible individuals.
To cite this abstract in AMA style:
Chow A, Koester S, Pepper-Tunick E, Lee P, Eckert M, Brenchley L, Gardner P, Li N, Schiffenbauer a, Volochayev R, Bayat N, McLean J, Rider L, Shenoi S, Stevens A, Dey N. Familial Clustering of Dysbiotic Oral and Fecal Microbiomes in Juvenile Dermatomyositis (JDM) [abstract]. Arthritis Rheumatol. 2023; 75 (suppl 9). https://acrabstracts.org/abstract/familial-clustering-of-dysbiotic-oral-and-fecal-microbiomes-in-juvenile-dermatomyositis-jdm/. Accessed .« Back to ACR Convergence 2023
ACR Meeting Abstracts - https://acrabstracts.org/abstract/familial-clustering-of-dysbiotic-oral-and-fecal-microbiomes-in-juvenile-dermatomyositis-jdm/