Session Information
Date: Friday, November 6, 2020
Title: Miscellaneous Rheumatic & Inflammatory Diseases I: Mechanisms of Disease (0459–0463)
Session Type: Abstract Session
Session Time: 10:00AM-10:50AM
Background/Purpose: Autosomal dominant hyper-IgE syndrome (AD-HIES), also known as Job’s syndrome, is a rare primary immunodeficiency caused by dominant-negative loss-of-function (LOF) mutations in signal transducer and activator of transcription 3 (STAT3). STAT3 LOF patients develop dermatitis, recurrent infections, and elevated serum IgE. Autoimmunity and auto-inflammation are common complication of primary immune deficiencies; however, systemic autoimmune features and the role of innate immune system are not well-characterized in AD-HIES.
Methods: A retrospective chart review of patients with STAT3 LOF seen at the National Institutes of Health was performed to identify patients who also had clinical and serologic features of systemic lupus erythematosus (SLE), as defined by American College of Rheumatology (ACR) 1997 classification criteria. To characterize lupus-associated innate immune dysregulation, type I interferon (IFN) stimulated genes (ISGs) were quantified by RT-PCR in neutrophils, in vitro neutrophil extracellular trap (NET) formation was measured by immunofluorescence, and serum anti-NET IgG autoantibodies were quantified by ELISA.
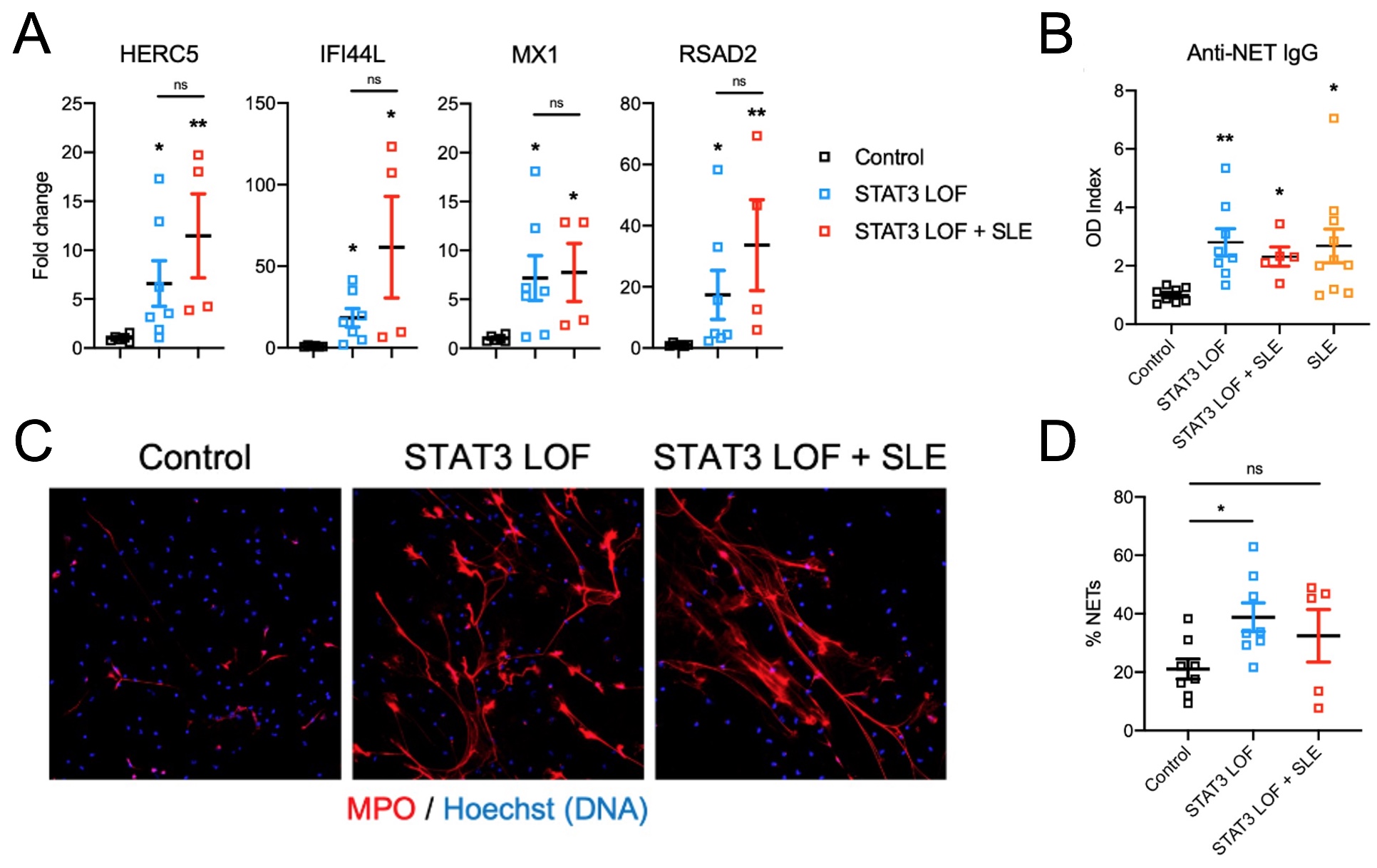
Results: We identified 10 out of 158 individuals (prevalence ~6.3%) with genetically confirmed STAT3 LOF mutations who developed systemic lupus erythematosus (SLE) or SLE-like manifestations, including immune complex glomerulonephritis (40%), cytopenias (50%), cutaneous disease (40%), and arthritis (40%). STAT3 LOF patients with SLE features were predominantly female (70%), and the onset of lupus and lupus-like disease features occurred between 12-24 years of age. All patients had positive ANA (100%), with a majority having anti-double-stranded DNA antibodies (90%), and hypocomplementemia (70%). Other lupus-associated-autoantibodies were also detected (Table 1). Immunosuppressive medications were required in 8 patients with systemic autoimmune features, which were well-tolerated and led to improvement in clinical symptoms. STAT3 LOF subjects with and without clinical features of SLE had increased expression of ISGs in neutrophils when compared to healthy controls. Increased spontaneous NET formation and anti-NET autoantibodies were observed in LOF STAT3 subjects, independent of the presence or absence of lupus-like symptoms and at higher levels than healthy controls (Figure 1).
Conclusion: Although autoimmunity is not a common finding in STAT3 LOF patients, we have identified SLE or SLE-like disease in about 6% of our cohort, with a high incidence of kidney involvement. These findings are associated with immune dysregulation characteristic of SLE including an increased IFN signature, NET formation, and various autoantibodies, including anti-NET antibodies. Significant immune dysregulation associated with dominant-negative STAT3 LOF mutations may explain the increased prevalence of systemic autoimmunity in these patients. Given this association, we recommend that patients with STAT3 LOF mutations be considered for rheumatology consultation for early recognition and treatment of SLE. The increased IFN signature also raises the potential for JAK-STAT modulation for therapy in these patients.
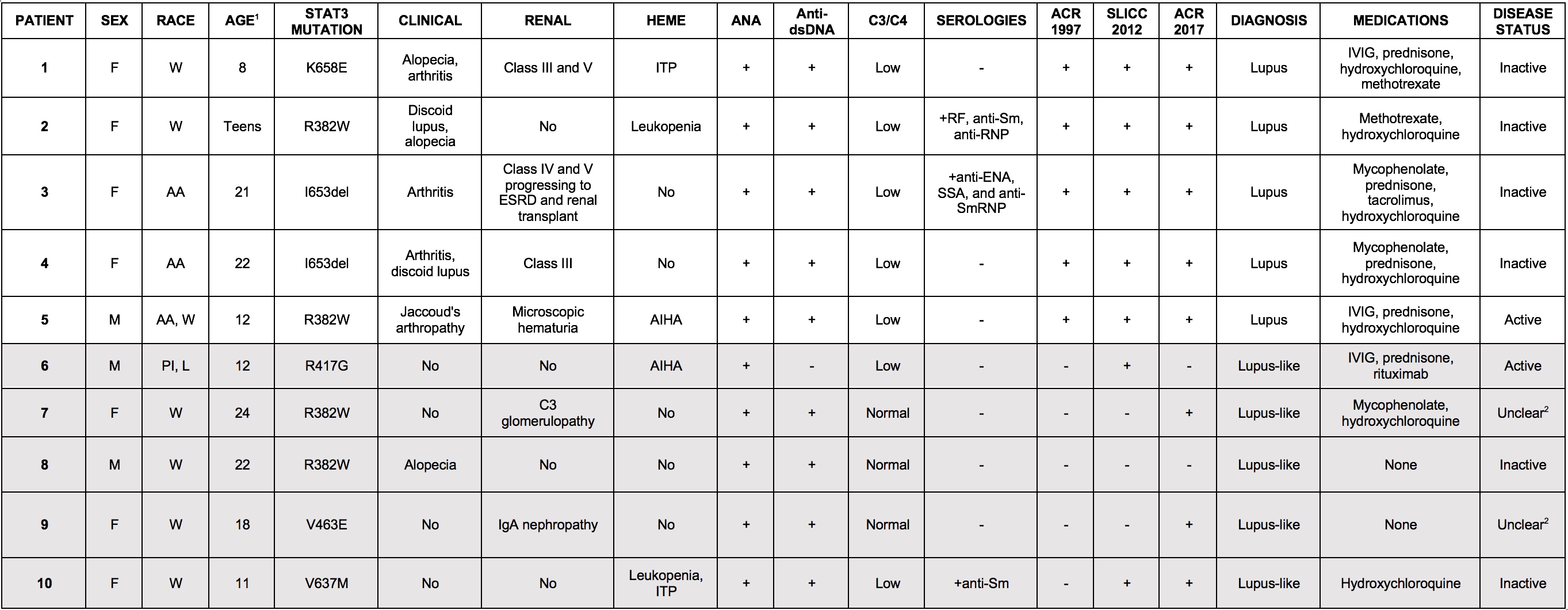 Table 1. Clinical characteristics of STAT3 LOF patients with lupus and lupus-like autoimmunity. Patients (n=10) with STAT3 LOF mutations were identified to have clinical and serologic features of lupus. Demographic information, disease manifestations, clinical laboratory data, and historic treatment protocols are shown. Diagnosis of lupus and lupus-like disease was made on the basis of fulfilling ACR 1997 classification criteria. Updated classification criteria for systemic lupus, SLICC 2012 and ACR 2017, are shown for comparison. Abbreviations are the following: W= white, AA= African-American, L= Latino, PI= Pacific Islander, ESRD= end-stage renal disease, ITP= idiopathic thrombocytopenic purpura, AIHA= autoimmune hemolytic anemia, IVIG= intravenous immunoglobulin, IVIG= intravenous immunoglobulin. 1Age at onset of lupus symptoms. 2Delineation of lupus and non-lupus symptoms is unclear at this time.
Table 1. Clinical characteristics of STAT3 LOF patients with lupus and lupus-like autoimmunity. Patients (n=10) with STAT3 LOF mutations were identified to have clinical and serologic features of lupus. Demographic information, disease manifestations, clinical laboratory data, and historic treatment protocols are shown. Diagnosis of lupus and lupus-like disease was made on the basis of fulfilling ACR 1997 classification criteria. Updated classification criteria for systemic lupus, SLICC 2012 and ACR 2017, are shown for comparison. Abbreviations are the following: W= white, AA= African-American, L= Latino, PI= Pacific Islander, ESRD= end-stage renal disease, ITP= idiopathic thrombocytopenic purpura, AIHA= autoimmune hemolytic anemia, IVIG= intravenous immunoglobulin, IVIG= intravenous immunoglobulin. 1Age at onset of lupus symptoms. 2Delineation of lupus and non-lupus symptoms is unclear at this time.
To cite this abstract in AMA style:
Dizon B, Goel R, Nakabo S, Urban A, Waldman M, Howard L, Darnell D, Buhaya M, Hasni S, Kaplan M, Freeman A, Gupta S. Lupus-like Autoimmunity and Increased Interferon Response in Patients with STAT3-deficient Hyper-IgE Syndrome [abstract]. Arthritis Rheumatol. 2020; 72 (suppl 10). https://acrabstracts.org/abstract/lupus-like-autoimmunity-and-increased-interferon-response-in-patients-with-stat3-deficient-hyper-ige-syndrome/. Accessed .« Back to ACR Convergence 2020
ACR Meeting Abstracts - https://acrabstracts.org/abstract/lupus-like-autoimmunity-and-increased-interferon-response-in-patients-with-stat3-deficient-hyper-ige-syndrome/