Session Information
Date: Monday, November 11, 2019
Title: Systemic Sclerosis & Related Disorders – Basic Science Poster
Session Type: Poster Session (Monday)
Session Time: 9:00AM-11:00AM
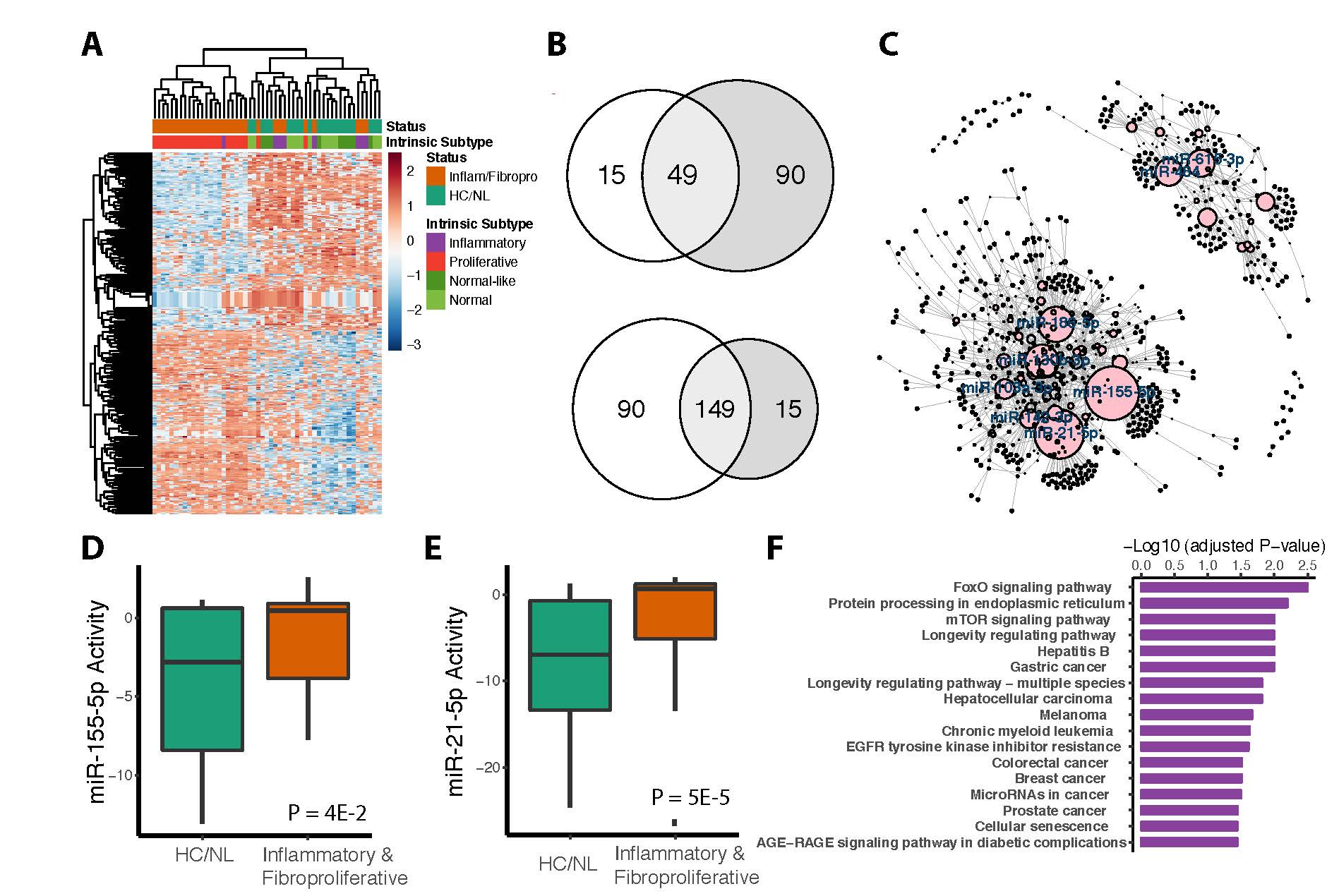
Background/Purpose: Systemic sclerosis (SSc; scleroderma) is a rare, heterogenous autoimmune disease characterized by skin fibrosis, vasculopathy and internal organ involvement. SSc patients can be divided into four intrinsic gene expression subtypes: inflammatory, fibroproliferative, limited and normal-like. The expression and activity of miRNAs, non-coding RNAs that post-transcriptionally regulate gene expression, have not been comprehensively characterized across the intrinsic gene expression subsets. We investigated the regulation and activity of miRNAs across the SSc subsets, developed a miRNA-target network, and experimentally validated a key hub of the network.
Methods: Gene expression profiles were downloaded for two patient cohorts, Milano et al. (GSE9285; 75 samples) and Assassi et al. (GSE58095; 102 samples). miRNA-target interactions (MTIs) were collected from miRTarBase; the activity of each miRNA is based on the expression of its target genes. The BASE algorithm was used to generate the miRNA regulatory activity scores for each miRNA in each sample. A Wilcoxon rank sum test was used to identify miRNAs with differential activity in inflammatory and fibroproliferative SSc samples. A miR-seq was performed on 8 SSc and 4 healthy control skin samples to validate findings. Targets strongly inhibited by cognate miRNAs were used to create the MTI network by calculating their negative correlation. The R package “igraph” was used for visualization and topological analysis of the network. Centrality measures were used to determine the node importance. For the most essential miRNAs, target genes were ranked and functionally annotated by pathway enrichment analysis using g:Profiler. miRNAs knockdowns in primary human SSc-differentiated macrophages were used to validate in-silico analyses.
Results: We identified a total of 198 miRNAs that were consistently differentially activated in both datasets (149 miRNAs were upregulated, and 49 miRNAs were downregulated). 1534 target genes were connected to these miRNAs in the network. Topological measurements of the network revealed that miR-155-5p and miR-21-5p had the broadest influence throughout the network. Both miR-155-5p and miR-21-5p showed consistently high activity in inflammatory and fibroproliferative SSc samples, and were highly expressed by miR-seq. MiR-155-5p has 92 target genes in this network that are significantly enriched in pathways that regulate inflammation and macrophage activation including the FoxO signaling pathway, which has been shown to mediate the prevention of autoimmunity.
Conclusion: We identified a list of miRNAs that show differential activity in SSc samples. MiR-155-5p and miR-21-5p were the most central in the regulatory network topologically, and have significantly higher activity and expression in inflammatory and fibroproliferative SSc samples. Over-activation of miR-155-5p leads to the loss of pathways associated with inhibition of autoimmune disease and proliferation of muscle cells.
Abstract_panel
To cite this abstract in AMA style:
Yuan Y, Wang Y, Pioli P, Whitfield M. Identification of miRNAs in Systemic Sclerosis Based on Activity and Network Analysis [abstract]. Arthritis Rheumatol. 2019; 71 (suppl 10). https://acrabstracts.org/abstract/identification-of-mirnas-in-systemic-sclerosis-based-on-activity-and-network-analysis/. Accessed .« Back to 2019 ACR/ARP Annual Meeting
ACR Meeting Abstracts - https://acrabstracts.org/abstract/identification-of-mirnas-in-systemic-sclerosis-based-on-activity-and-network-analysis/