Session Information
Session Type: Poster Session A
Session Time: 10:30AM-12:30PM
Background/Purpose: Recent single-cell RNA sequencing (scRNA-seq) studies of the rheumatoid arthritis (RA) synovium have highlighted the heterogeneity of cell states present during active disease. It is unclear how these diverse cell types interact with one another in situ, and the role of synovial microenvironments in determining cell states is not well understood. Sequencing-based spatial transcriptomics (ST) is an innovative technology that provides high-throughput molecular profiling of tissue sections while preserving spatial information. In this work, we use ST and a scalable unsupervised machine learning method using Dirichlet variational autoencoders (DirVAEs) to identify cellular communities and map previously-defined cell types to these microenvironments.
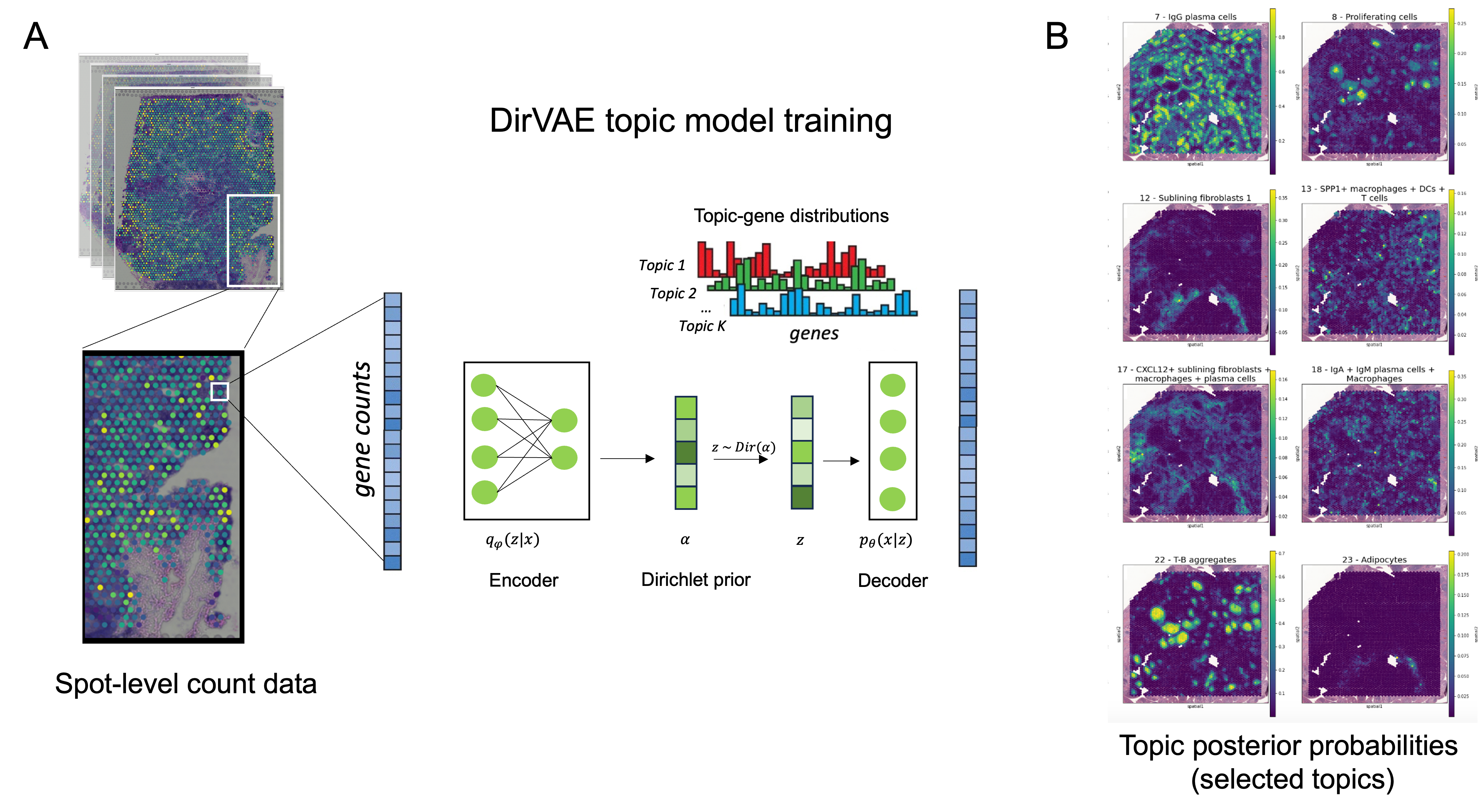
Methods: We applied the 10X Visium CytAssist workflow to 8 synovial tissue sections from 6 patients meeting EULAR 2010 criteria. All synovial sections contained numerous discrete lymphoid aggregates. These 8 ST datasets were aggregated, and one DirVAE topic model was trained on the combined dataset (Fig 1A). The generated latent features, which can represent individual or spatially co-occurring cell types, are referred to as “topics”, analogous to topics in standard latent Dirichlet allocation (LDA). We identified discriminative genes and assigned the cell type(s) contained in each topic based on inference on published annotated RA scRNA-seq data. Cell type assignments were also confirmed using pathway analysis of the top ranked genes in each topic.
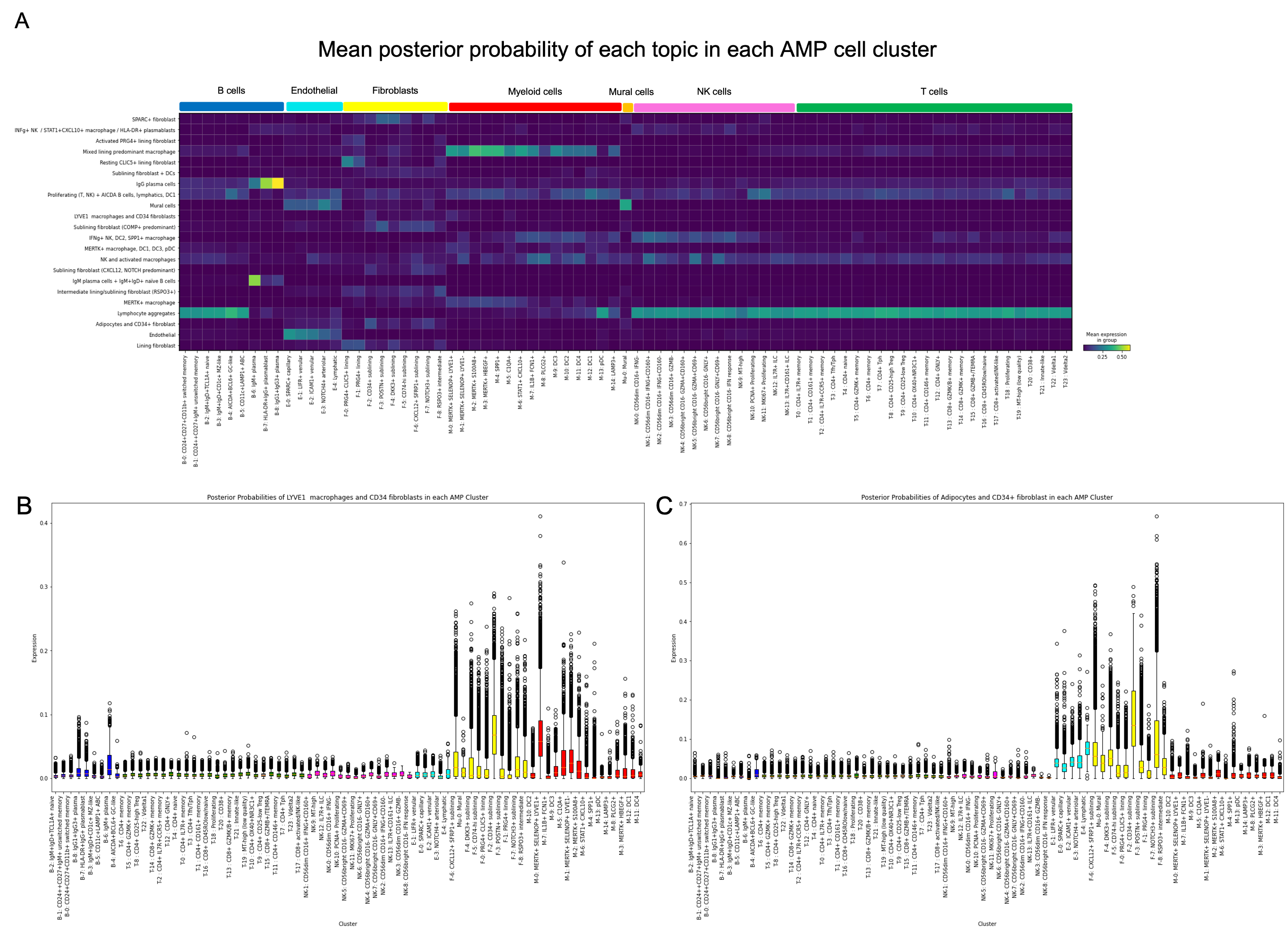
Results: Our model successfully identified topics that were found to correspond to cell types that are specific to RA, such as lining and sublining fibroblasts, plasma cells, T and B lymphocytes, myeloid cells, and endothelial cells (Fig 2). Most topics were found to contain a combination of cell types that may interact with one another given their spatial proximity. Notably, CD34+ progenitor-like fibroblasts co-occurred in separate topics with (1) LYVE1+ macrophages and (2) adipocytes (Fig 2). Pathway analysis highlighted the predicted role of complement across all topics containing fibroblasts except for the topic containing SPARC+ collagen-producing fibroblasts. Notably, though all tissues were consistent with a lymphoid pathotype, there was significant variation in the occurrence of each topic across tissue sections, indicating significant differences in the spatial architecture including cell composition and cell-cell interactions between tissues (Fig 3).
Conclusion: Our DirVAE-based deconvolution method resolved fine-grained cell states from ST data and provided information beyond simple cell type deconvolution. Our model has allowed detection of possible interactions between previously-defined cell types and allowed us to map the variation of these cellular communities across multiple tissues. This variation, even across tissues similarly prominent lymphoid aggregates, highlights the need to further investigate spatial heterogeneity in the RA synovium to decipher the pathophysiology of RA.

To cite this abstract in AMA style:
Periyakoil P, Smith M, Kshirsagar M, Ramirez D, Dicarlo E, Goodman S, Donlin L, Leslie C. Deep Topic Modeling Deconvolves Cell States in Spatial Transcriptomic Profiles of Rheumatoid Arthritis Synovial Tissue [abstract]. Arthritis Rheumatol. 2024; 76 (suppl 9). https://acrabstracts.org/abstract/deep-topic-modeling-deconvolves-cell-states-in-spatial-transcriptomic-profiles-of-rheumatoid-arthritis-synovial-tissue/. Accessed .« Back to ACR Convergence 2024
ACR Meeting Abstracts - https://acrabstracts.org/abstract/deep-topic-modeling-deconvolves-cell-states-in-spatial-transcriptomic-profiles-of-rheumatoid-arthritis-synovial-tissue/