Session Information
Session Type: Poster Session B
Session Time: 9:00AM-10:30AM
Background/Purpose: The detection of anti-citrullinated protein antibodies (ACPA) is associated with increased risk for development of future Rheumatoid Arthritis (RA). The biological events that underpin the loss of immune tolerance and the progression toward pathogenic autoimmunity remain unclear. We sought to identify changes in circulating cytokine/chemokine networks that are associated with the presence and persistence of ACPA, and with the progression towards inflammatory arthritis in an at-risk cohort of first-degree relatives (FDR) of RA patients.
Methods: We studied baseline serum samples from a cohort of at-risk FDR (n = 147) and quantified serum cytokines/chemokines using a 48-plex platform. Longitudinal outcomes were defined on the basis of ACPA status and progression into inflammatory arthritis (IA). K-means clustering, differential expression, and principal components analysis were used to identify group level differences. A co-expression module analysis was used to identify enriched networks. ACPA Fab glycosylation was calculated as a % of total IgG. A Random Forest (RF) machine learning approach was used to classify ACPA positive individuals. Network analysis was used to understand underlying biological processes based on protein expression.
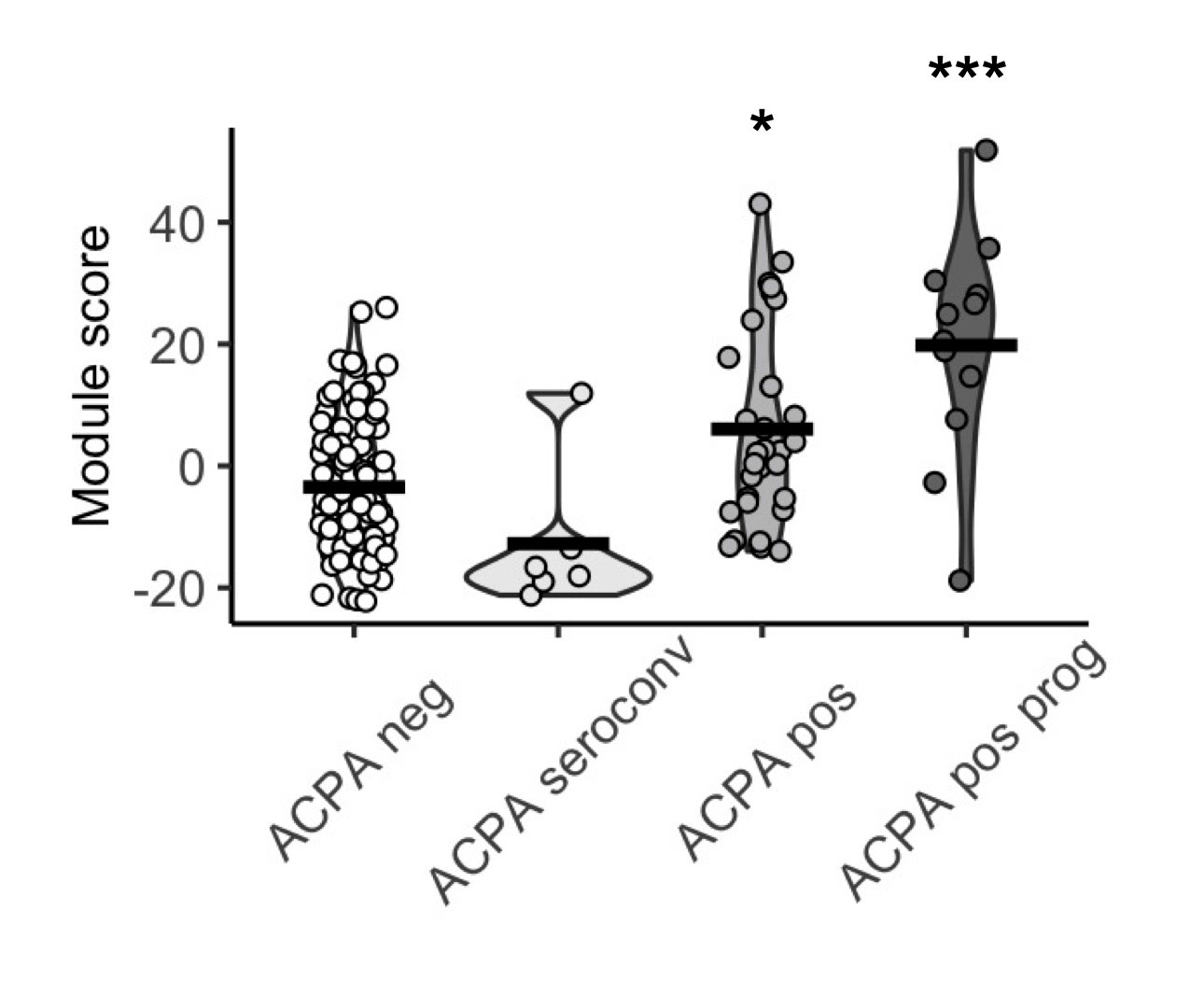
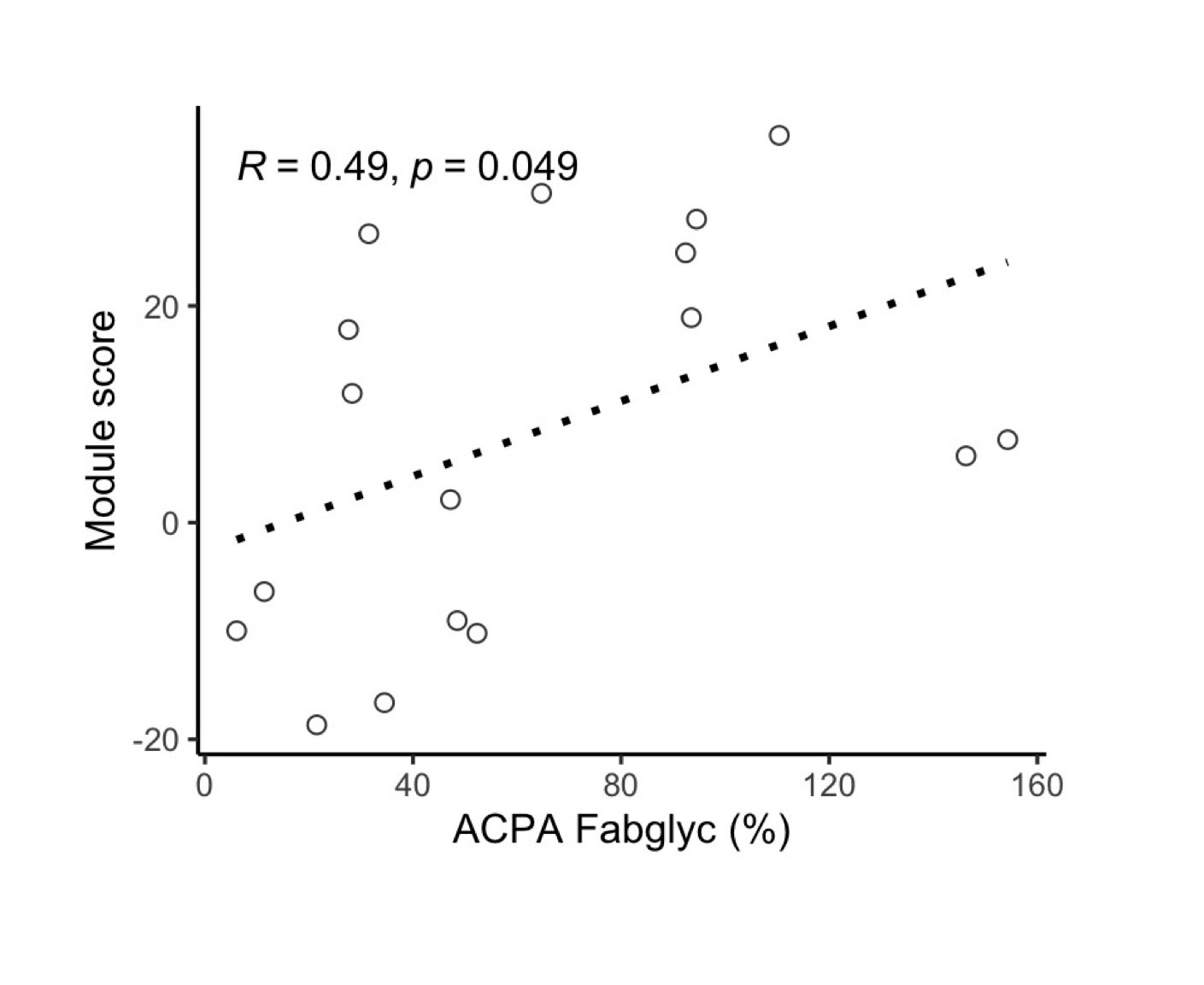
Results: Baseline demographics were balanced between ACPA positive (n = 48) and ACPA negative FDR (n = 99). We defined 6 proteomic clusters, with enrichment of ACPA positive samples in cluster 6 (p < 0.0001). As such, 23 of 24 differentially expressed proteins in ACPA positive samples were upregulated, including CCL11, IFNg and IL1b. A co-expression network was identified, and protein network member expression (n = 23 members) was higher in baseline ACPA positive sera (p = 0.0005) and in individuals who progressed into IA (p = 0.0003), but not those who subsequently became seronegative for ACPA during the observation period. The co-expression network correlated with ACPA Fab glycosylation % (R = 0.49, p = 0.049), a known marker of future disease progression. RF achieved an area under the curve of 0.767 to classify ACPA positive sera in a test dataset. Network analysis revealed upregulation of JAK-STAT signalling in those at highest risk to develop future IA, along with several drugs that may modulate this pathway.
Conclusion: A serum proteomic analysis focused on cytokine/chemokine networks provides a rich dataset with which to better understand biological processes in pre-clinical RA. This approach allowed us to identify key pathways that were associated with ACPA seropositivity, and progression towards IA onset. A combination of ACPA and the identified serum biomarkers, may increase the precision with which to predict future RA in at-risk individuals.
To cite this abstract in AMA style:
O'Neil L, MENG X, McFadyen C, Fritzler M, El-Gabalawy H. Serum Proteomic Networks Associate with Pre-clinical Rheumatoid Arthritis Autoantibodies and Longitudinal Outcomes [abstract]. Arthritis Rheumatol. 2022; 74 (suppl 9). https://acrabstracts.org/abstract/serum-proteomic-networks-associate-with-pre-clinical-rheumatoid-arthritis-autoantibodies-and-longitudinal-outcomes/. Accessed .« Back to ACR Convergence 2022
ACR Meeting Abstracts - https://acrabstracts.org/abstract/serum-proteomic-networks-associate-with-pre-clinical-rheumatoid-arthritis-autoantibodies-and-longitudinal-outcomes/