Session Information
Session Type: Poster Session B
Session Time: 9:00AM-10:30AM
Background/Purpose: Current treatments for SLE do not adequately prevent disease progression. This lack of efficacy, in part, relates to the molecular heterogeneity of disease. The objective of this study was to use DNA methylation from flaring SLE patients to identify distinct biological subtypes of SLE associated with disease activity
Methods: Fifty-three SLE flaring patients recruited during routine clinical care were studied. A flare was defined by the treating rheumatologist and characterized with the SLEDAI score. Blood samples, clinical data and medication use were collected at the time of flare and approximately three months later. Genome-wide methylation profiles were generated using Illumina’s Infinium Human MethylationEPIC BeadChip. Quantile normalization and background subtraction with dye-bias normalization were performed using minfi and methylation sites (CpGs) with high detection p-values, cross-reactive probes, and those measuring genetic variants were excluded. Differentially methylated positions (DMPs) associated with SLEDAI score were identified using limma. Models accounted for the paired design and adjusted for blood cell proportions, batch, age, sex, medications, and genetic principal components. Consensus hierarchical clustering was then performed on the 5,000 most significant DMPs using methylation M-values from the highest SLEDAI visit to identify patient subgroups. Clinical features and DMPs unique to each subgroup were identified using ANOVA, chi-square, and limma. Gene ontology enrichment analysis was performed on the set of DMPs unique to each subtype.
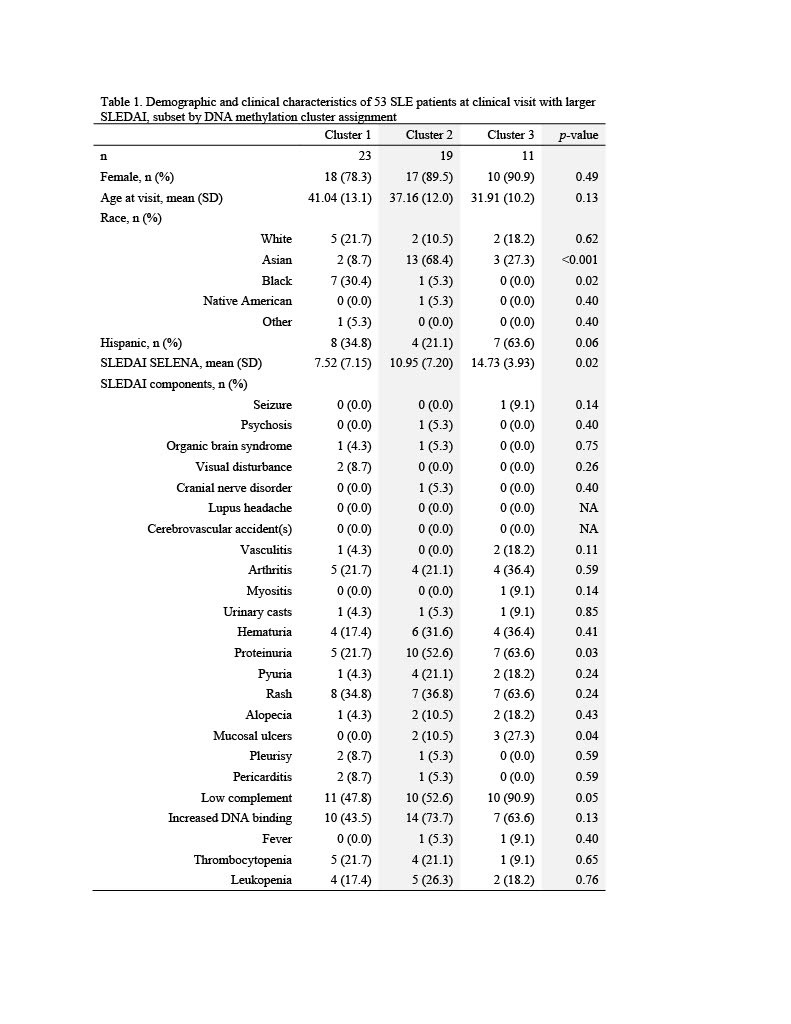
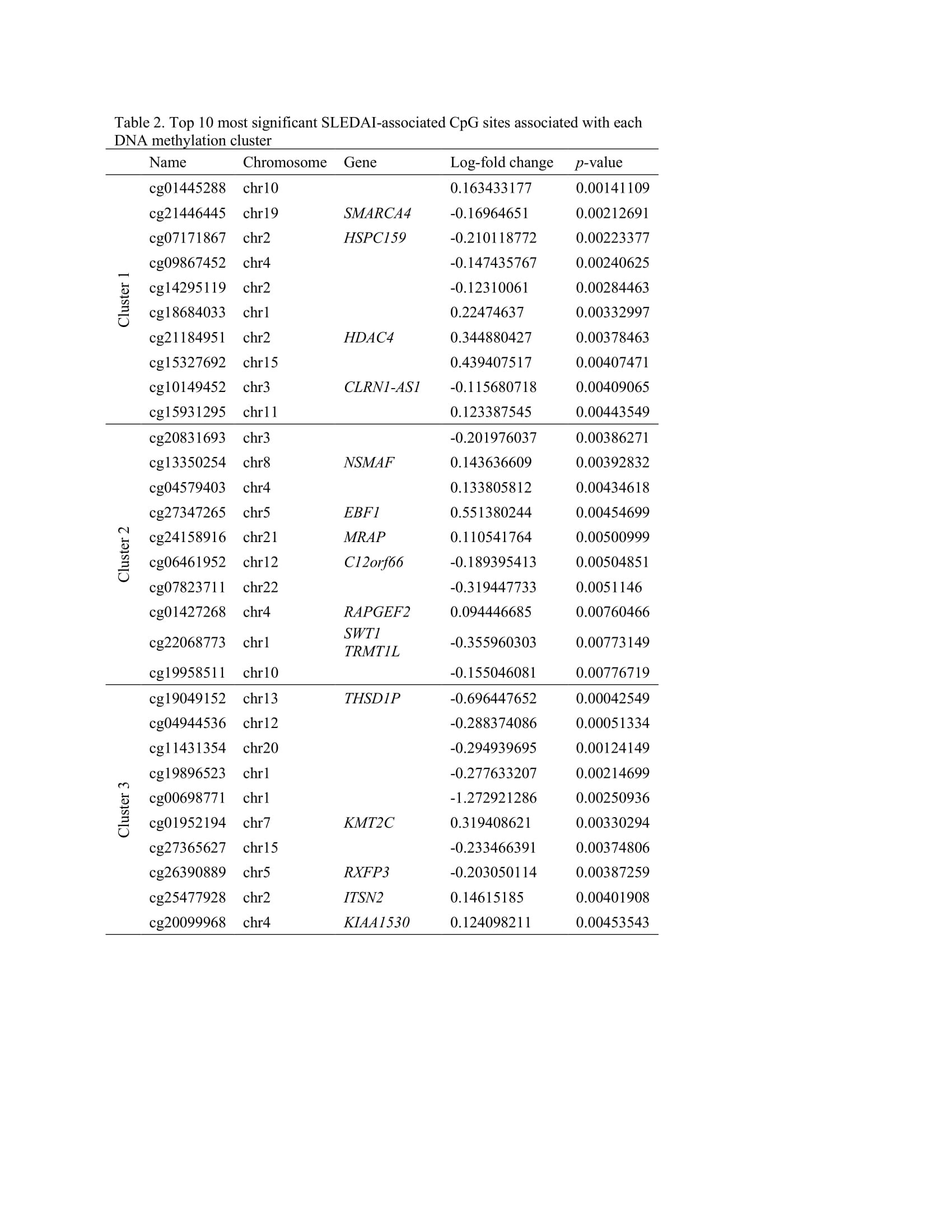
Results: The mean SLEDAI score among flaring samples was 10.2 (sd=6.3). Three clusters of patients were identified using DNA methylation data (Table 1). Mean SLEDAI was lowest for Cluster 1 (7.5 (sd=7.2)) and increased for each subsequent group (11.0 (sd=7.2) and 14.7 (sd=3.9) for Cluster 2 and Cluster 3, respectively). Cluster 2 was characterized by a higher percentage of Asians (68%) and Cluster 3 a higher percentage of Hispanics (64%). More than 100 DMPs were identified as unique for each cluster (p< 0.05). The most significant gene ontology pathways for Cluster 1 were predominantly phosphorylation processes and regulation of defense response by viruses. For Cluster 2, notable DMPs were found in EBF1 (B-cell transcription factor), C12orf66 (regulator of mTOR signaling), and SWT1 (riboendonuclease expressed in basophils). Top pathways included import into the nucleus and adrenergic receptor signaling. For Cluster 3, notable DMPs include KMT2C (methyltransferase expressed in neutrophils), RFXP3 (associated with monocyte recruitment and differentiation), ITSN2 (regulator of clathrin-coated vesicles and induction of T cell antigen receptor endocytosis), and KIAA1530 (gene involved in UV-sensitive syndrome). Pathways for Cluster 3 included synaptic transmission and several neuronal membrane pathways (Tables 2, 3).
Conclusion: Three biologically distinct subgroups of flaring SLE patients were identified using DNA methylation data. This subtyping might be used to better inform treatment decisions and targeted therapies based on relevant underlying biological pathways.

To cite this abstract in AMA style:
Horton M, Nitithalm J, Taylor K, Trupin L, Katz P, Yazdany J, Dall'Era M, Barcellos L, Criswell L, M Lanata C. Subsetting SLE Patients Based on DNA Methylation at the Time of Disease Flare [abstract]. Arthritis Rheumatol. 2022; 74 (suppl 9). https://acrabstracts.org/abstract/subsetting-sle-patients-based-on-dna-methylation-at-the-time-of-disease-flare/. Accessed .« Back to ACR Convergence 2022
ACR Meeting Abstracts - https://acrabstracts.org/abstract/subsetting-sle-patients-based-on-dna-methylation-at-the-time-of-disease-flare/