Session Information
Date: Monday, November 9, 2020
Title: SLE – Diagnosis, Manifestations, & Outcomes Poster III: Bench to Bedside
Session Type: Poster Session D
Session Time: 9:00AM-11:00AM
Background/Purpose: Cross-sectional studies have shown associations between DNA methylation differences in whole blood with Systemic Lupus Erythematosus (SLE) status and outcomes such as lupus nephritis. However, DNA methylation is not static and a proportion of DNA methylation changes in response to environmental cues and with the effects of time. To study the trajectory of DNA methylation in whole blood in SLE we examined 101 participants at two time points in a well-characterized SLE cohort.
Methods: 101 participants from the California Lupus Epidemiology Study (CLUES), a longitudinal cohort of individuals with rheumatologist-confirmed SLE were studied. Participants are followed every 2 years through in-person research visits. DNA extracted from blood at cohort enrollment and after 2 years was analyzed on the illumina EPIC Beadchip. Single nucleotide polymorphism (SNP) genotype data was generated on the Affymetrix LAT1 World Array. First we performed a paired t-test between methylation at cohort enrollment and 2 years after, after adjusting for cell composition and methylation plate. We then applied a mixed linear model on methylation values adjusting for age, sex, population stratification and cell proportions. We previously reported 256 differentially methylated CpGs according to disease severity at cohort enrollment.1 We performed focused analysis on these 256 CpGs as well as exploratory genome-wide analysis.
1Lanata et al, Nat Comm 10, 3902 (2019)
Results: After quality control 806,000 CpGs were analyzed. Of the 256 CpGs previously associated with disease severity, 55 CpGs (21.4%) had a significant change in methylation between the 2 time points (p < 2E-04, effect size > 0.03, paired t-test), 52 with a decrease and 3 with an increase in methylation. The largest effect size was observed for cg13452062 in IFI44L with a mean decrease of 0.16 (p=6.4e-25) and the most represented genes were IFI44L, IFITM1, PARP9 with 4 CpGs each. In the mixed linear model, the 55 CpGs remained significant (p < 2E-04) although we found a significant effect of cell proportions with changes in methylation. As representative examples, we find a significant effect of CD8 proportions with cg07285983 in RAPGAP1L, B cell proportions with cg13452062 in IFI44L and monocyte proportions with cg10959651 in RDAD2 (Table 1). Disease activity ((SLE Disease Activity Index score) did not vary significantly between time 1 and time 2 (p=1.0, paired t-test). Exploratory genome-wide analyses found 151 CpGs (0.02%) with a significant change in methylation between the 2 time points (p < 6.2E-08, effect size > 0.03 paired t-test). Of these, 55 (33%) were in the previously described 256 CpGs. Of the remaining CpGs, top associations included CpGs in TLR6, RSAD2, MSRA and COPS8.
Conclusion: In this study, a small proportion of DNA methylation signatures in blood previously associated with SLE phenotypes fluctuated over time. These changes in methylation associate with circulating cell proportion in SLE patients. Additional longitudinal studies will be needed to further describe and validate candidate CpGs that represent stable biomarkers of disease state as well as CpGs that change in relationship to cell proportions and other unmeasured factors.
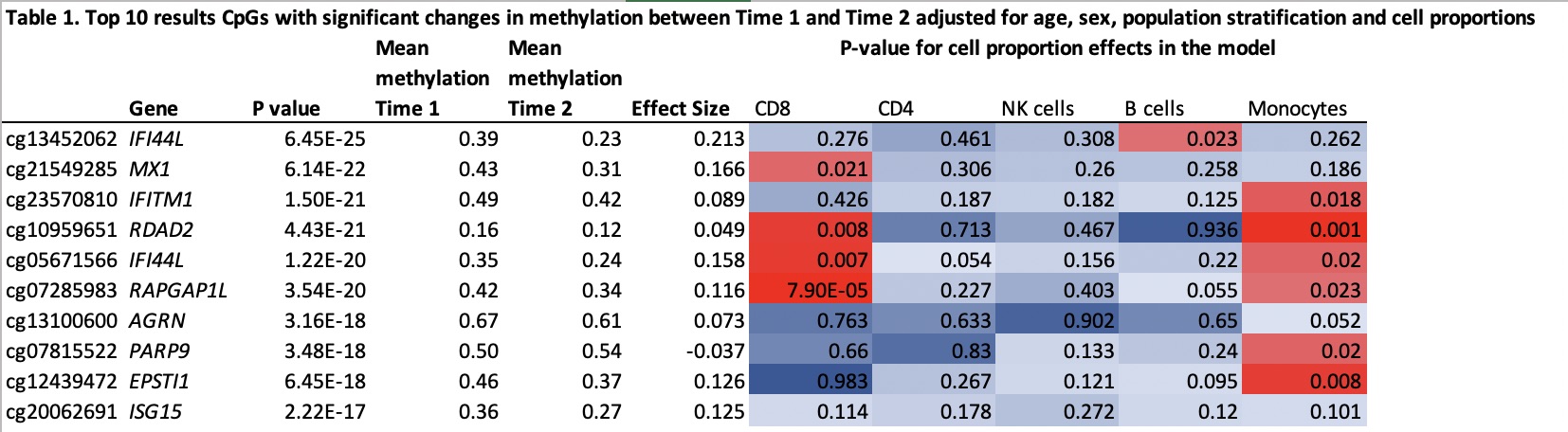 Top 10 results CpGs with significant changes in methylation between Time 1 and Time 2 adjusted for age, sex, population stratification and cell proportions
Top 10 results CpGs with significant changes in methylation between Time 1 and Time 2 adjusted for age, sex, population stratification and cell proportions
To cite this abstract in AMA style:
Lanata C, Nititham J, Taylor K, Chung S, Trupin L, Katz P, Dall'Era M, Yazdany J, Sirota M, Barcellos L, Criswell L. Longitudinal Blood DNA Methylation in a Multi-ethnic Cohort of SLE Patients [abstract]. Arthritis Rheumatol. 2020; 72 (suppl 10). https://acrabstracts.org/abstract/longitudinal-blood-dna-methylation-in-a-multi-ethnic-cohort-of-sle-patients/. Accessed .« Back to ACR Convergence 2020
ACR Meeting Abstracts - https://acrabstracts.org/abstract/longitudinal-blood-dna-methylation-in-a-multi-ethnic-cohort-of-sle-patients/