Session Information
Session Type: Poster Session C
Session Time: 8:30AM-10:30AM
Background/Purpose: S1PR1 signaling plays critical roles in EC barrier function and vascular health, but until now, research on S1PR1 modulation in autoimmune rheumatic disease has been focused on lymphocyte trafficking. Importantly, endothelial cells have never been targeted for the treatment of autoimmune rheumatic disease. In prior work, we showed that S1PR1 modulators attenuated immune complex (IC) mediated vascular injury in skin and lung. In vitro, S1PR1 agonists rescued EC barrier function and decreased loss of VE-cadherin expression in human umbilical vein ECs (HUVEC) challenged with IC activated neutrophils. In this study we asked whether EC S1PR1 signaling limits experimental arthritis and whether it does so, at least in part, by limiting VE-cadherin shedding.
Methods: Mice with an EC specific S1PR1 KO or gain of function (GOF) were subjected to K/BxN serum induced arthritis (SIA) by injecting 100 µl of serum days 0 and 2. To determine how pharmacologic modulation of S1PR1 signaling affected SIA, WT animals were treated with S1PR1 agonist, CYM-5442 or antagonist NIBR-0213 30 mg/kg or vehicle controls. Clinical scores were obtained daily after day 2 in a blinded manner and mice were sacrificed on days 2-3 or on day 8. Synovial fluids from arthritic ankles obtained on days 2-3 were assessed by ELISA and western blot for soluble VE-cadherin. H&E-stained paraffin sections were scored by a pathologist in a blinded manner. HUVECs treated with S1PR1 inhibitor NIBR-0213 or vehicle control were assessed by western blot.
Results: S1PR1 ECKO mice had augmented arthritis at earlier time points and mice with EC S1PR1 GOF had delayed disease compared to controls (n=7-8 animals/group, p< 0.05; Fig 1). Similarly, the S1PR1 agonist CYM-5442 delayed the onset of arthritis (n= 7 mice/group p< 0.05; Fig 2), while an S1PR1 antagonist NIBR induced earlier and more severe disease (n=4 animals/group p= 0.05). Synovial fluid from S1PR1 ECKO mice showed increased VE-cadherin shedding (n= 6; p ≤ 0.01) and Evans blue extravasation (n=6; p= 0.1) at early time points after SIA compared to controls, suggesting that increased vascular permeability via VE-cadherin shedding contributed to arthritic damage. S1PR1 ECKO mice also showed increased VE-cadherin shedding in their bronchoalveolar lavage fluid at baseline, suggesting that S1PR1 signaling inhibits VE-cadherin at homeostasis. To determine whether blockade of S1PR1 signaling induce VE-cadherin shedding in vitro, we treated HUVEC with NIBR-0213 (Fig 3). There was a marked increase in VE-cadherin shedding which was blocked by non-specific metalloprotease inhibitor marimastat (MM). Human RA single cell RNA seq data in ECs from the Accelerated Medicine Partnership (AMP) show that expression of EC S1PR1 and EC Sphingosine kinase 1, a key enzyme for the production of the endogenous ligand, S1P, are both statistically significantly lower than in OA controls.
Conclusion: Our data reveal that S1PR1 signaling restrains VE-cadherin shedding and maintains EC vascular barrier function and thereby attenuates arthritis. Augmenting EC S1PR1 signaling is a potential novel, non-immunosuppressive approach to treat inflammatory arthritis.
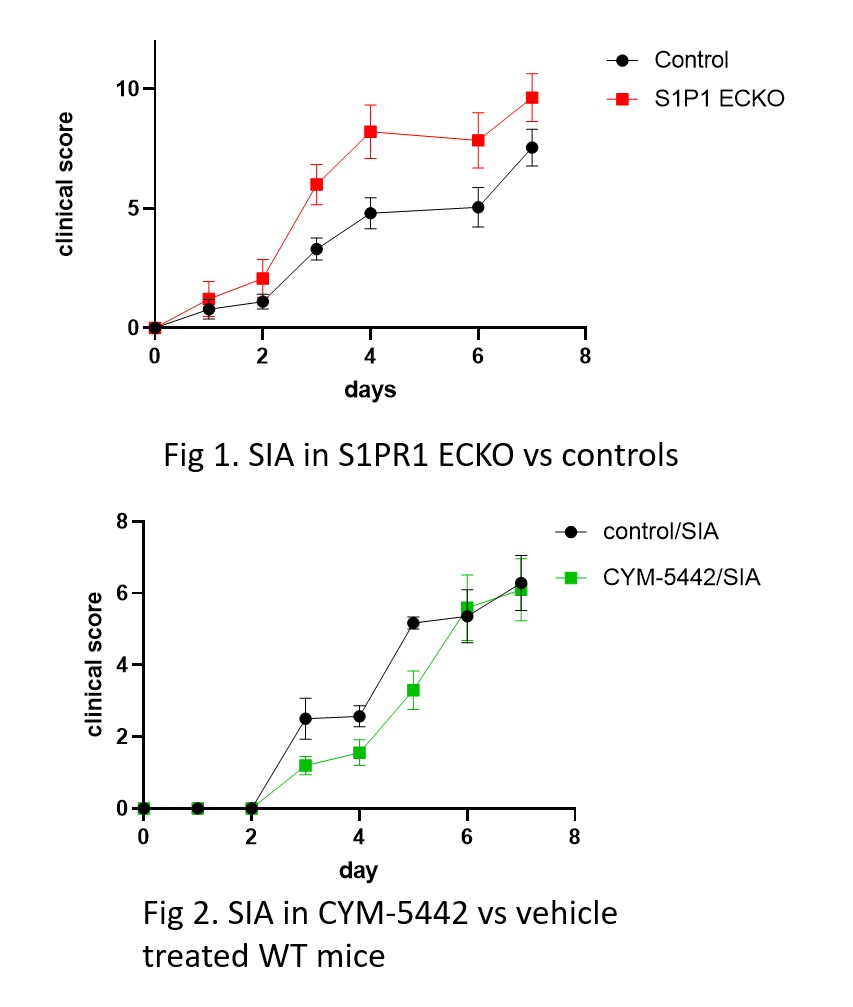 Fig 1. S1PR1 ECKO mice show increased inflammatory injury in response to SIA Fig 2. The S1PR1 agonist CYM-5442 delays SIA in WT mice
Fig 1. S1PR1 ECKO mice show increased inflammatory injury in response to SIA Fig 2. The S1PR1 agonist CYM-5442 delays SIA in WT mice
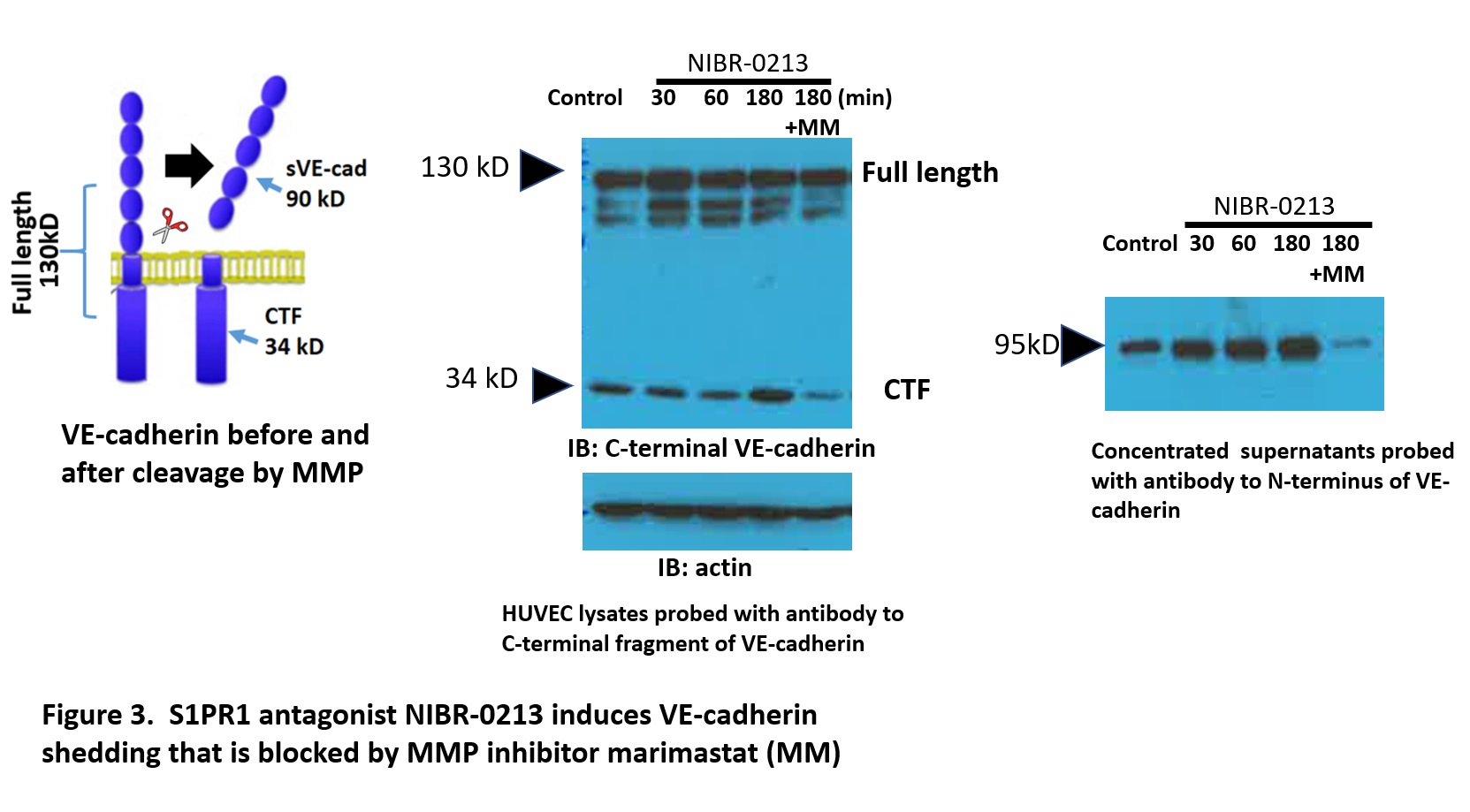 S1PR1 antagonist NIBR-0213 induces shedding of VE-cadherin in HUVEC that is blocked by the MMP inhibitor Marimastat (MM)
S1PR1 antagonist NIBR-0213 induces shedding of VE-cadherin in HUVEC that is blocked by the MMP inhibitor Marimastat (MM)
To cite this abstract in AMA style:
Burg N, Malpass R, Blobel C, Salmon J. Enhancing Endothelial Cell Barrier Function via Sphingosine -1 Phosphate Receptor 1 (S1PR1) – a Novel Treatment for Experimental Arthritis [abstract]. Arthritis Rheumatol. 2021; 73 (suppl 9). https://acrabstracts.org/abstract/enhancing-endothelial-cell-barrier-function-via-sphingosine-1-phosphate-receptor-1-s1pr1-a-novel-treatment-for-experimental-arthritis/. Accessed .« Back to ACR Convergence 2021
ACR Meeting Abstracts - https://acrabstracts.org/abstract/enhancing-endothelial-cell-barrier-function-via-sphingosine-1-phosphate-receptor-1-s1pr1-a-novel-treatment-for-experimental-arthritis/