Session Information
Date: Sunday, November 17, 2024
Title: Systemic Sclerosis & Related Disorders – Basic Science Poster I
Session Type: Poster Session B
Session Time: 10:30AM-12:30PM
Background/Purpose: Cardiac fibrosis is a common complication in Systemic sclerosis (SSc), but the pathogenesis is largely unknown. We have previously shown that monocytes isolated from scleroderma patients have low levels of the transcription factor Fli1, and that this conferred profibrotic properties with increased collagen deposition in coculture with cardiac fibroblasts, independent of TGFβ pathway activation. In this study, we further investigated the mechanism that contributes to these profibrotic effects.
Methods:
CD14+ beads were used to isolate monocytes from controls and SSc patients, and peritoneal and cardiac macrophages where isolated from mice with myeloid specific deletion of Fli1 (LysMCre/Fli1fl/fl), crossed with Tdtomato/GFP reporter mice. siRNA was used to downregulate Fli1 and galectin-3 (Gal-3) in primary human monocyte derived macrophages, and effects where assessed separately or in co-culture with primary human cardiac fibroblasts. Rapamycin was used to block the mTOR complex 1 (mTORC1) pathway. Immunofluorescence staining with specific antibodies or western blot was used to assess protein levels as indicated. QuPath was used to quantify immunofluorescent staining. Picrosirius red staining was used for collagen quantification in the cardiac tissue. ChIP-qPCR were used to determine the transcriptional regulation of Gal-3 (galectin-3) by Fli1.
Results:
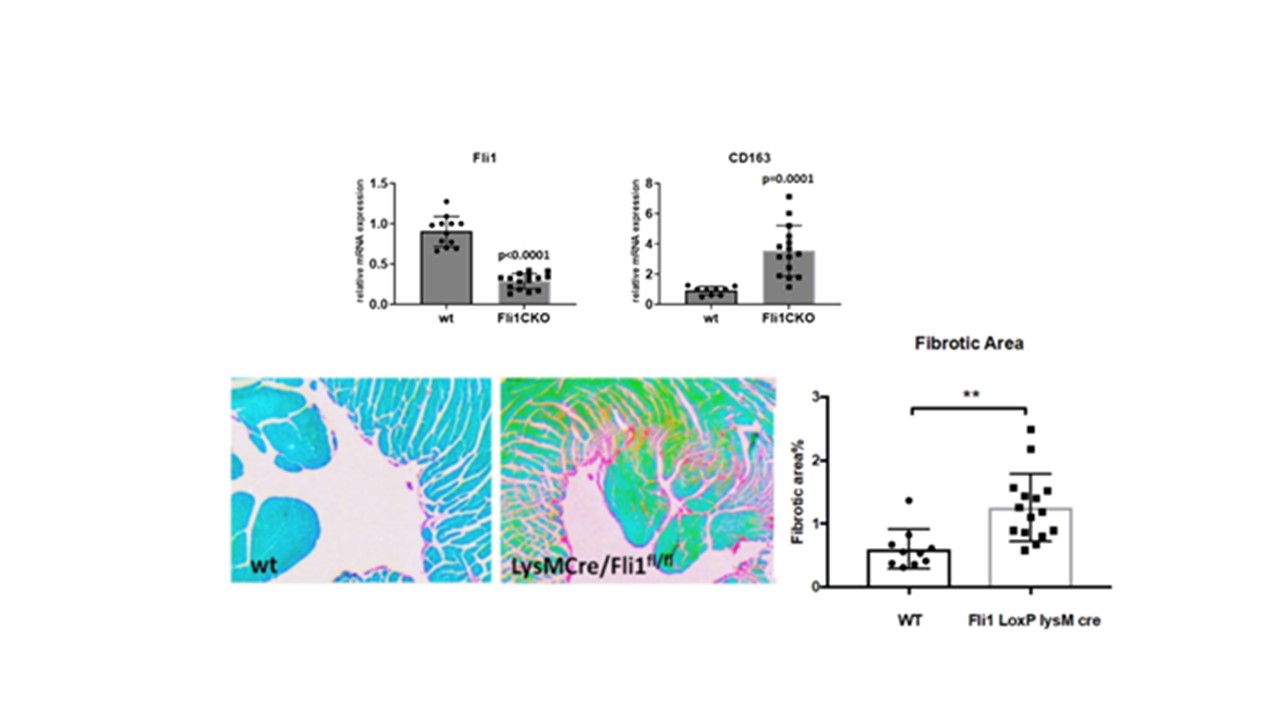
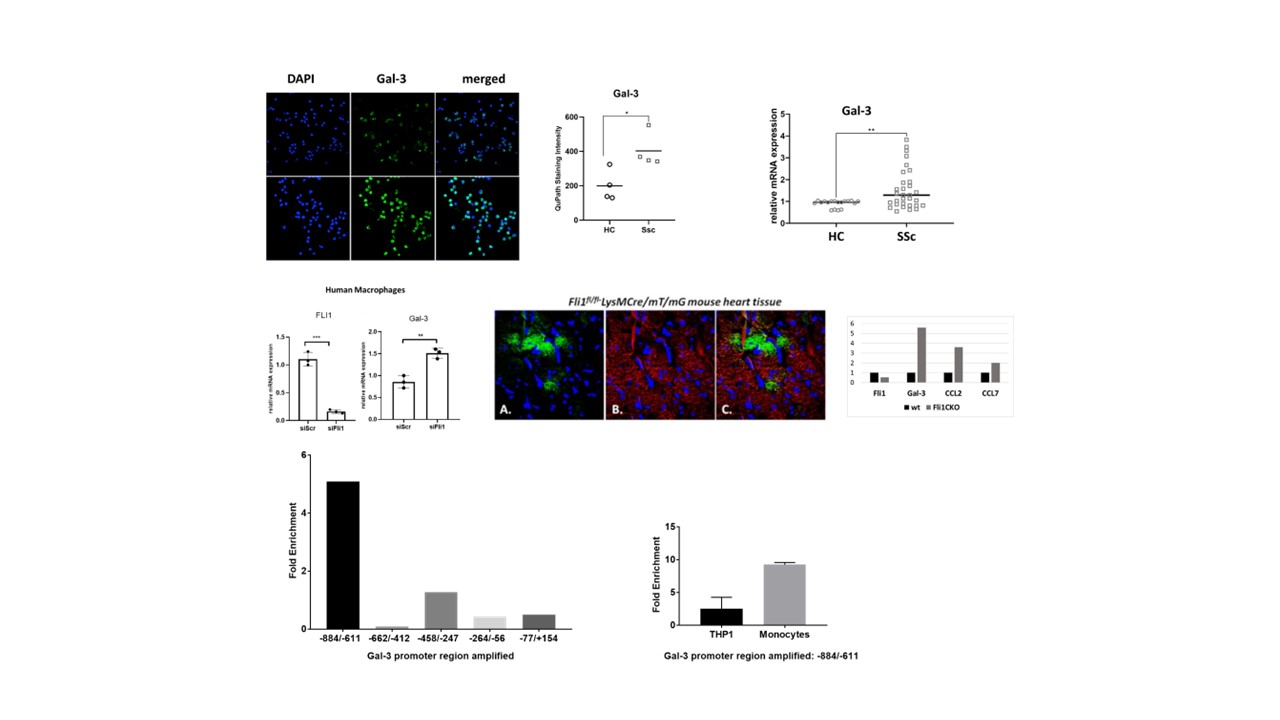
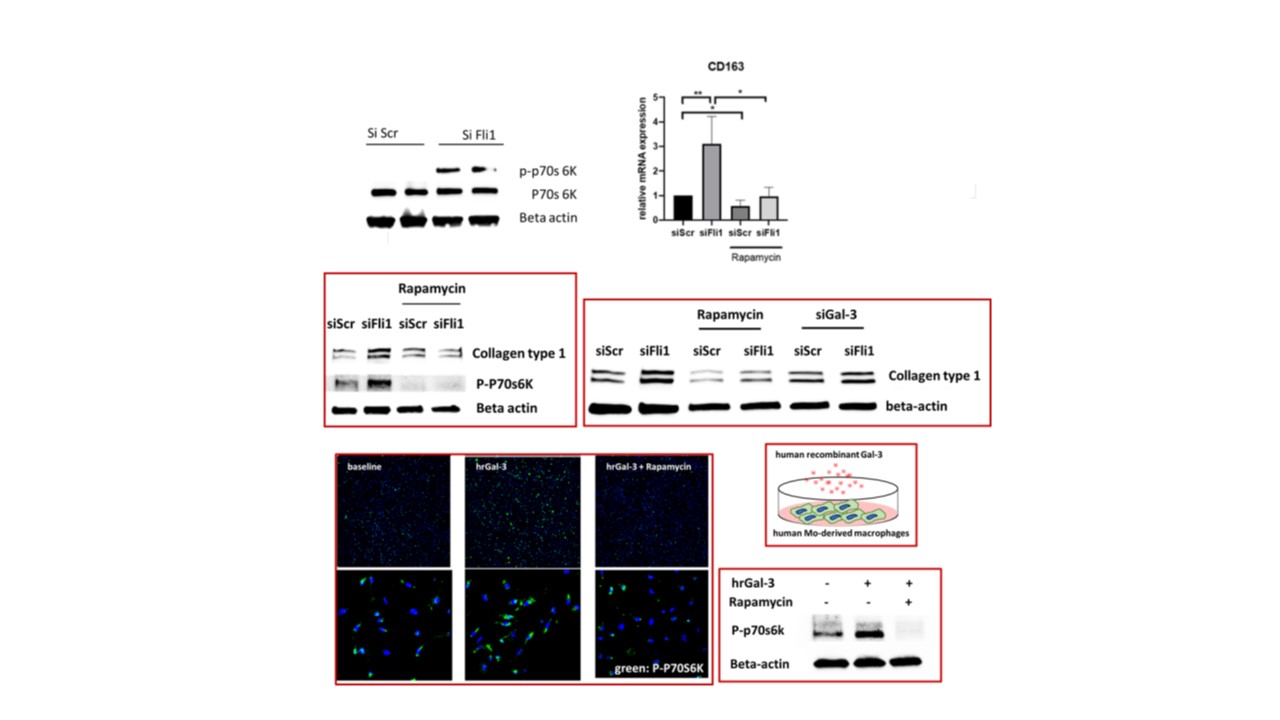
Mice with conditional knockout of Fli1 in myeloid cells (LysMCre/Fli1fl/fl) had increased cardiac collagen deposition (Fig.1). Depletion of Fli1 in human myeloid cells resulted in upregulation of Galectin-3 (Gal-3), a profibrotic β-galactoside-binding lectin secreted by alternatively-activated macrophages that was recently linked to SSc fibrosis. Additionally, Gal-3 was overexpressed in SSc monocytes (Fig 2). Enhanced Fli1 binding to the LGALS3 promoter region -884 to -611 was seen in human macrophages, and macrophages isolated from Fli1fl/flLysMCre/mT/mG mice had elevated Gal-3 expression. Co-culture of Fli1 silenced myeloid cells with cardiac fibroblasts led to activation of the mTORC1 in both cell types, as evidenced by the increased P-p70S6K. Addition of human recombinant Gal-3 activated mTORC1 signaling in both macrophages and fibroblasts. Rapamycin prevented the profibrotic effects seen with low myeloid Fli1 and depletion of Gal-3 in myeloid cells prevented collagen upregulation in fibroblasts.
Conclusion:
Decreased Fli1 in SSc myeloid cells leads to upregulation and increased secretion of Gal-3 in the extracellular space, resulting in activation of mTORC1, profibrotic activation of macrophages and collagen deposition by fibroblasts. Inhibiting the mTOR/Gal-3 pathway could serve as a therapy targeted at myeloid cell dysfunction and cardiac fibrosis in scleroderma.
To cite this abstract in AMA style:
El adili F, Mudhibadhi M, ligresti G, Trojanowska M, Bujor A. Downregulated Fli1 in Scleroderma Myeloid Cells Contributes to Cardiac Fibrosis via a Galectin-3/mTOR Dependent Pathway [abstract]. Arthritis Rheumatol. 2024; 76 (suppl 9). https://acrabstracts.org/abstract/downregulated-fli1-in-scleroderma-myeloid-cells-contributes-to-cardiac-fibrosis-via-a-galectin-3-mtor-dependent-pathway/. Accessed .« Back to ACR Convergence 2024
ACR Meeting Abstracts - https://acrabstracts.org/abstract/downregulated-fli1-in-scleroderma-myeloid-cells-contributes-to-cardiac-fibrosis-via-a-galectin-3-mtor-dependent-pathway/