Session Information
Date: Monday, October 27, 2025
Title: (1147–1190) Miscellaneous Rheumatic & Inflammatory Diseases Poster II
Session Type: Poster Session B
Session Time: 10:30AM-12:30PM
Background/Purpose: Heterozygous loss-of-function mutations in CTLA4 cause a spectrum of immune dysregulation, characterized by hypogammaglobulinemia, autoimmune cytopenias, and lymphoproliferation, yet penetrance and expressivity remain highly variable across cohorts[1-3] To characterize the clinical, genetic, and therapeutic features of a prospective Chinese cohort with CTLA-4 haploinsufficiency, and to compare their management outcomes with those reported in the largest international series.
Methods: Seven patients diagnosed by whole-exome sequencing underwent systematic evaluation of clinical phenotypes, laboratory parameters, imaging and histopathology, and treatment responses. Data were contrasted against the global 133-patient cohort described by Kuehn et al[1]
Results: All individuals harbored heterozygous CTLA4 variants—four missense and three truncating mutations—exhibiting classical features of immune dysregulation alongside marked phenotypic heterogeneity, even among family members with identical genotypes Relative to the international cohort’s median diagnostic delay exceeding 10 years, our patients reached genetic confirmation within 2–5 years, enabling earlier initiation of targeted therapy. Nearly all received abatacept (CTLA-4-Ig), achieving sustained disease stabilization and tapering of corticosteroids, complemented by IVIG to correct hypogammaglobulinemia Unlike the 9% malignancy rate observed globally—often EBV-associated—no tumors emerged during 6–12 months of follow-up, suggesting that earlier intervention may mitigate oncogenic risk. Proactive family screening identified asymptomatic carriers, underscoring the value of genetic counseling for at-risk relatives.
Conclusion: This study demonstrates that early genetic screening coupled with prompt abatacept-based immunomodulation and IVIG replacement can markedly shorten diagnostic latency, stabilize multi-system disease, and potentially reduce malignancy complications in CTLA-4 haploinsufficiency. These findings provide a model for precision-medicine approaches in diverse populations.[1] Schwab C, Gabrysch A, Olbrich P, et al. Phenotype, penetrance, and treatment of 133 cytotoxic T-lymphocyte antigen 4–insufficient subjects[J/OL]. Journal of Allergy and Clinical Immunology, 2018, 142(6): 1932-1946. DOI: 10.1016/j.jaci.2018.02.055.[2] Jamee M, Hosseinzadeh S, Sharifinejad N, et al. Comprehensive comparison between 222 CTLA‐4 haploinsufficiency and 212 LRBA deficiency patients: a systematic review[J/OL]. Clinical and Experimental Immunology, 2021, 205(1): 28-43. DOI: 10.1111/cei.13600.[3] Schubert D, Bode C, Kenefeck R, et al. Autosomal-dominant immune dysregulation syndrome in humans with CTLA4 mutations[J/OL]. Nature Medicine, 2014, 20(12): 1410-1416. DOI: 10.1038/nm.3746.
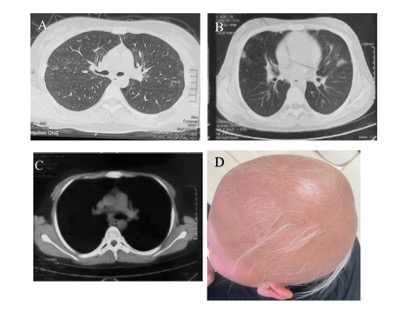 Fig1.Typical clinical manifestations of subjects with CTLA-4 deficiency.
Fig1.Typical clinical manifestations of subjects with CTLA-4 deficiency.
A. P1: Multiple ground-glass nodules in the lungs
B. P6: Lung inflammation
C. P6:Mediastinal lymphadenopathy
D. P5: Complete baldness of the head and eyebrows, after treatment, the hair grows back into vellus hair
.jpg) Table1 : Clinical and genetic features of Chinese patients with CTLA-4h
Table1 : Clinical and genetic features of Chinese patients with CTLA-4h
.jpg) Table2:Comparison of this cohort with the largest existing cohort
Table2:Comparison of this cohort with the largest existing cohort
To cite this abstract in AMA style:
Liu G, zhang J, Xu j, Dai J, Shen M. Clinical and Genetic Features of CTLA-4 Haploinsufficiency : A Prospective Study in China [abstract]. Arthritis Rheumatol. 2025; 77 (suppl 9). https://acrabstracts.org/abstract/clinical-and-genetic-features-of-ctla-4-haploinsufficiency-a-prospective-study-in-china/. Accessed .« Back to ACR Convergence 2025
ACR Meeting Abstracts - https://acrabstracts.org/abstract/clinical-and-genetic-features-of-ctla-4-haploinsufficiency-a-prospective-study-in-china/