Session Information
Date: Monday, November 14, 2022
Title: SLE – Animal Models Poster
Session Type: Poster Session D
Session Time: 1:00PM-3:00PM
Background/Purpose: Neurologic dysfunction occurs in up to 80% of SLE patients. Cognitive impairment is one of the most frequent manifestations of neuropsychiatric SLE (NPSLE). There are multiple mechanisms for cognitive impairment, including cytokines, type I interferon, HMGB1, antibodies, and medications. We have shown that antibodies (Ab) to double-stranded (ds) DNA that cross-react with the N-methyl D-aspartate receptor (NMDAR), which we have termed DNRAbs, are present in ~30% of SLE patients. In a mouse model, we demonstrated that DNRAbs cause acute brain injury in the CA1 hippocampus through an excitotoxic mechanism and chronic neuronal injury with microglial activation and loss of neuronal spines and dendrites. We hypothesized that microglial activation after DNRAb penetration into the hippocampus may depend on the interaction of HMGBA1 with the RAGE receptor. To test this hypothesis, we studied the neuropathological consequences of exposure to DNRAb or control Ab in RAGE deficient (KO) and WT C57BL/6 mice.
Methods: RAGE KO and WT C57BL/6 mice were immunized to produce DNRAbs and then given lipopolysaccharide (LPS) to afford the Abs access to the brain. After two weeks of LPS administration, stereological techniques were used to evaluate acute neuronal loss in the CA1 region of the hippocampus. Eight weeks after LPS administration, Golgi-stained neurons were analyzed to assess dendritic complexity. Also, using super-resolution con-focal microscopy, IBA-1 labeled microglial cells were evaluated for activation by assessment of morphology and presence of CD68 according to standard protocols.
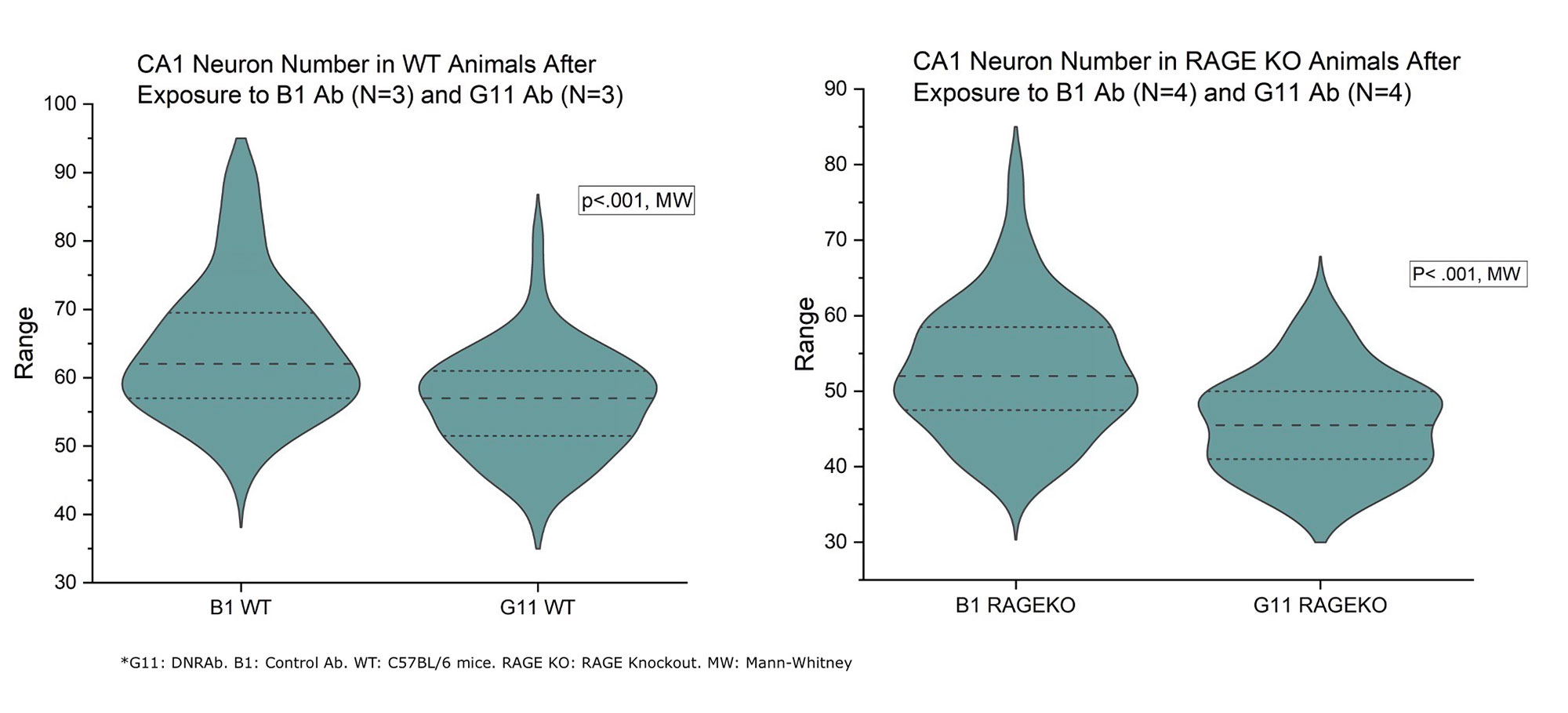
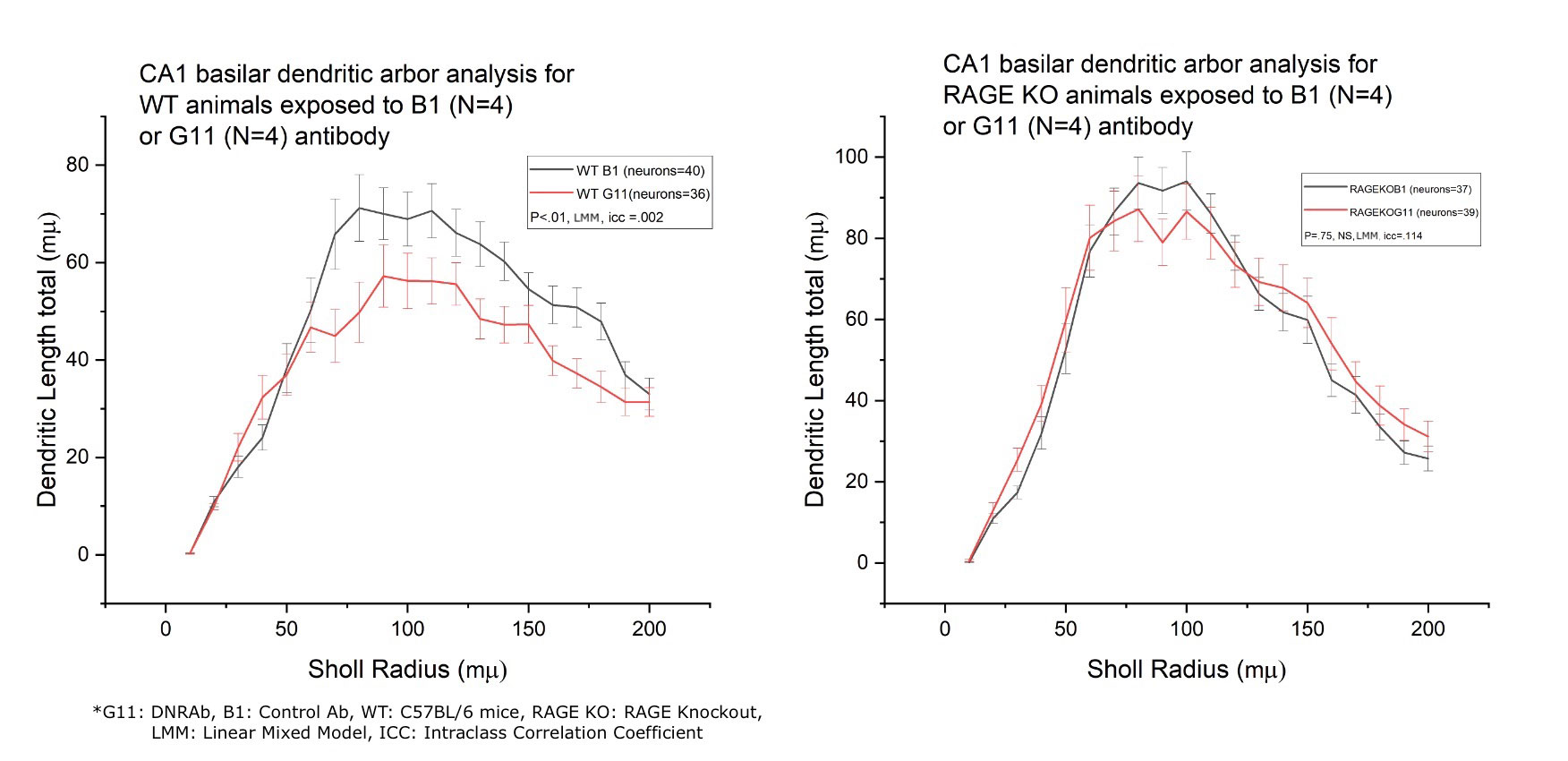
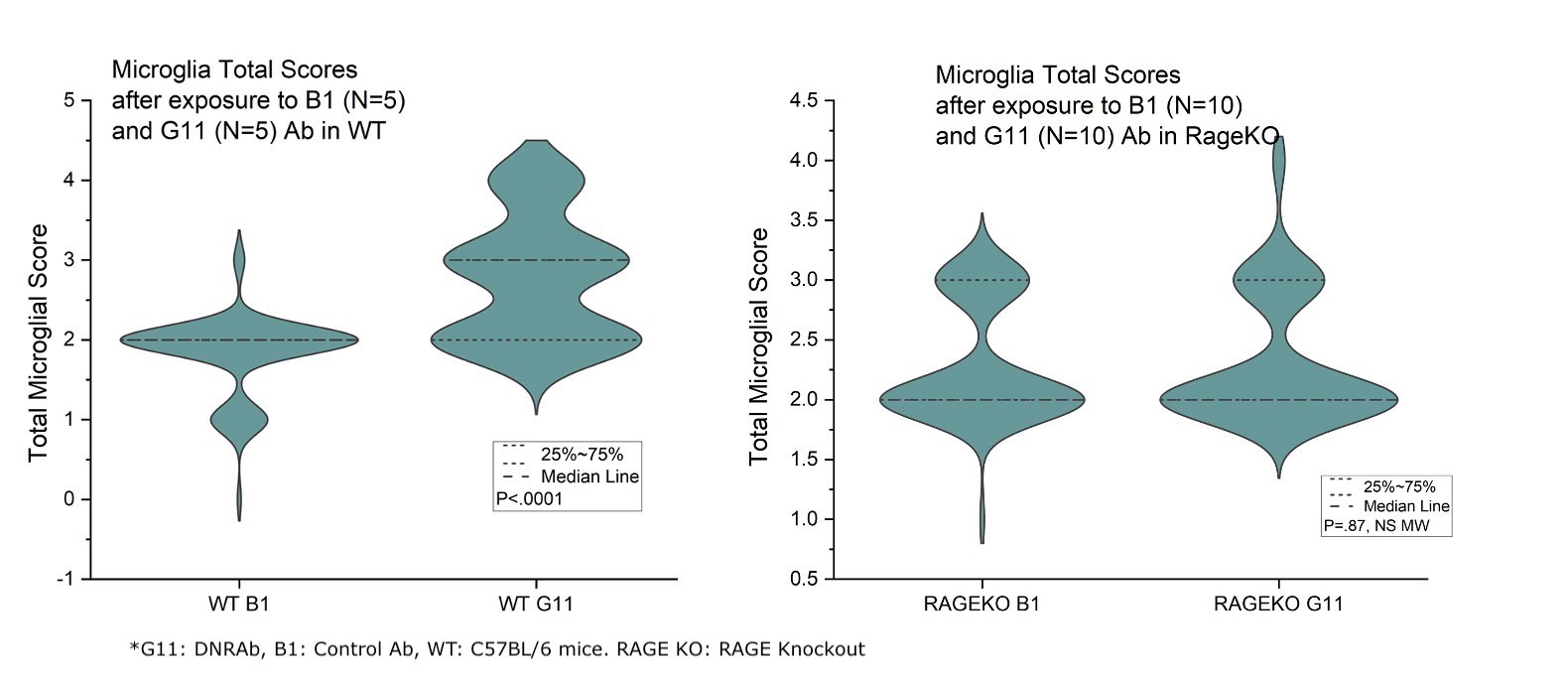
Results: This study revealed that DNRAb significantly decreased the number of neurons in the CA1 region of the hippocampus in both WT C57BL/6 and RAGE KO mice (p< 0.001) two weeks after LPS administration compared to the control antibody (Figure-1). Further, eight weeks later, as expected, Golgi-stained neurons showed that DNRAb caused a significant neuronal dendritic loss in the WT C57BL/6 mice compared to control Ab (p< 0.01). However, there was no significant difference in dendritic arborization (p=0.75) between DNRAb and control Ab exposed in RAGE KO mice (Figure-2). Although microglial activation was increased in the DNRAb exposed WT C57BL/6 mice (p< 0.001), DNRAb exposed RAGE- KO mice demonstrated comparable microglial activation with the control Ab exposed mice (Figure-3).
Conclusion: These findings suggest that exposure to DNRAb mediates acute excitotoxic neuronal loss in the hippocampus, irrespective of RAGE; however, the preservation of dendritic integrity and the absence of microglial activation in RAGE-KO mice strongly suggests that RAGE is a key mediator of the chronic neurotoxic effects of DNRAbs. These findings prompt further study of the role of RAGE in NSPLE patients.
To cite this abstract in AMA style:
Toz B, wingard J, Volpe B, Diamond B. A Role for the RAGE Receptor in Chronic Neurotoxicity After Exposure to SLE Antibodies That Cross React with dsDNA and the NMDA Receptor [abstract]. Arthritis Rheumatol. 2022; 74 (suppl 9). https://acrabstracts.org/abstract/a-role-for-the-rage-receptor-in-chronic-neurotoxicity-after-exposure-to-sle-antibodies-that-cross-react-with-dsdna-and-the-nmda-receptor/. Accessed .« Back to ACR Convergence 2022
ACR Meeting Abstracts - https://acrabstracts.org/abstract/a-role-for-the-rage-receptor-in-chronic-neurotoxicity-after-exposure-to-sle-antibodies-that-cross-react-with-dsdna-and-the-nmda-receptor/