Session Information
Session Type: Abstract Submissions (ACR)
Background/Purpose: DNA methylation is a basic mechanism involved in epigenetic regulation that affects gene transcription by the addition of a methyl group to the cytosine residue within a CpG dinucleotide to form methylated cytosine. The objective of this work is to identify and analyze the genome-wide DNA methylation profiles of human articular chondrocytes from a population-based case-control study of OA.
Methods: DNA methylation profiling was performed using the Infinium HumanMethylation27 beadchip (Illumina Inc.), which allows interrogation of 27,578 highly informative CpG loci. Previously, cartilage isolated DNA from 23 OA patients and 19 healthy controls was bisulfite-modified, using the EZ DNA methylation kit (Zymo Research) and hybridized according to the manufacturerxs instructions. DNA methylation b-values were normalized using GenomeStudio v3.0 (Illumina Inc.). Appropriate bioinformatics analyses were carried out using both R bioconductor software packages and Babelomics suite v 4.2 (babelomics.bioinfo.cipf.es).
Results: A first approach based on an unsupervised clustering method for the most variable CpG loci (n=508) showed three distinct groups of samples called cluster 1, cluster 2 and cluster 3. Specifically, cluster 2 formed a particularly tight cluster with a characteristic DNA methylation profile (Figure 1). The analyses of the biological relevance of the differentially methylated genes in cluster 2 compared with non-cluster 2 by means of a gene set enrichment approach, showed that some of the biological processes significantly altered were those related to cellular adhesion, morphogenesis/angiogenesis and regulation of cell proliferation, all of them hypermethylated in cluster 2; on the contrary, those genes related to cytokine secretion/production as well as immune response and inflammation appeared significantly hypomethylated in cluster 2.
Conclusion: The genome-wide methylation analysis shows a clearly distinct epigenetic profile for OA patients. The DNA methylation profile could be one of the reasons of the existence of different forms OA and could also be related to both the prevalence and progression of this disease.
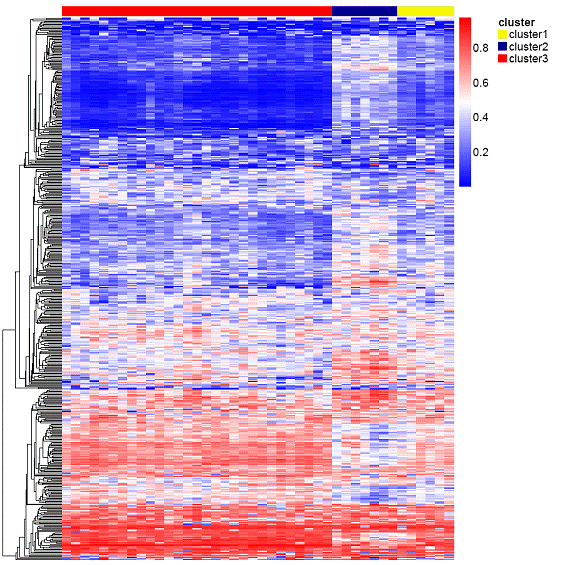
Figure 1. Heatmap showing the three distinct clusters. Cluster 2 shows a characteristic DNA methylation profile clearly different from the other two clusters.
Disclosure:
I. Rego-Pérez,
None;
J. Fernandez-Tajes,
None;
M. Fernandez-Moreno,
None;
M. Tamayo Novas,
None;
A. Mosquera Rey,
None;
N. Oreiro,
None;
C. Fernandez-Lopez,
None;
J. L. Fernandez Garcia,
None;
F. J. Blanco,
None.
« Back to 2012 ACR/ARHP Annual Meeting
ACR Meeting Abstracts - https://acrabstracts.org/abstract/a-genome-wide-dna-methylation-analysis-reveals-different-methylation-patterns-in-the-oa-disease/