Session Information
Date: Sunday, October 26, 2025
Title: (0593–0640) Systemic Lupus Erythematosus – Diagnosis, Manifestations, & Outcomes Poster I
Session Type: Poster Session A
Session Time: 10:30AM-12:30PM
Background/Purpose: Individuals with systemic lupus erythematosus (SLE) experience significant variability in clinical outcomes. Our current understanding of the causes of disease heterogeneity are limited, preventing successful risk stratification. To investigate the contribution of epigenetic variation to disease heterogeneity, we generated DNA methylation profiles for a large cohort of SLE patients and examined methylation patterns associated with presence of nephritis, given its importance to morbidity and mortality in SLE.
Methods: We studied 1,882 multi-ancestry SLE patients recruited during routine clinical care. Clinical data were collected at study enrollment and peripheral blood cell collection. Genome-wide methylation profiles were generated using Illumina’s EPICv2 BeadChip which includes 937,690 CpG sites. Samples were randomly subset into discovery (n= 1505; ~80%) and validation (n= 377; ~20%) cohorts. Normalization and quality control were performed using minfi. Methylation sites (CpGs) with high detection p-values and cross-reactive probes were excluded. Among the discovery cohort, differentially methylated positions (DMPs) associated with nephritis were identified using limma. We used methylation beta values for coefficient interpretation, and methylation M values to determine statistical significance. Models adjusted for genetic principal components, Houseman estimated blood cell proportions, batch, age, disease duration, and sex. DMPs with FDR q< 0.05 were considered significant. Significant DMPs were then tested for their association in the validation cohort using the same models. DMPs with FDR q < 0.05 and the same direction of effect in both cohorts were considered validated.
Results: In the discovery cohort, we identified 5,783 CpGs with FDR q< 0.05. Of these, 12 CpGs had at least a 5% difference in methylation between nephritis cases and non-nephritis SLE controls. 184 CpGs had FDR q < 0.05 and the same direction of effect in the validation cohort. Of these validated CpGs, 13 had at least a 5% difference in methylation between nephritis cases and controls. Validated CpGs included CpGs in IL1R1 (receptor for Interleukin-1), STAB1 (a scavenger receptor expressed in macrophages and modulator of inflammation), and TRIM62 (a tripartite motif involved in innate immunity and antiviral response). Of the validated CpG sites, the absolute change in methylation between nephritis cases and controls ranged from 0.2 to 9.5%. A majority of these were hypomethylated; 13 were located within genes, and 23 were in promoter regions. Gene set enrichment analysis revealed enrichment in T-cell production and lymphocyte mediated immunity, although adjusted p-values were not significant (Figure 2).
Conclusion: In the largest SLE multi-ancestry methylation data set to date, we identified 5,783 CpGs that were differentially methylated in SLE patients with nephritis, and we validated approximately 3% of these CpGs in a validation cohort. These results suggest that methylation patterns may provide opportunities to further subtype SLE patients for risk stratification in clinical settings.
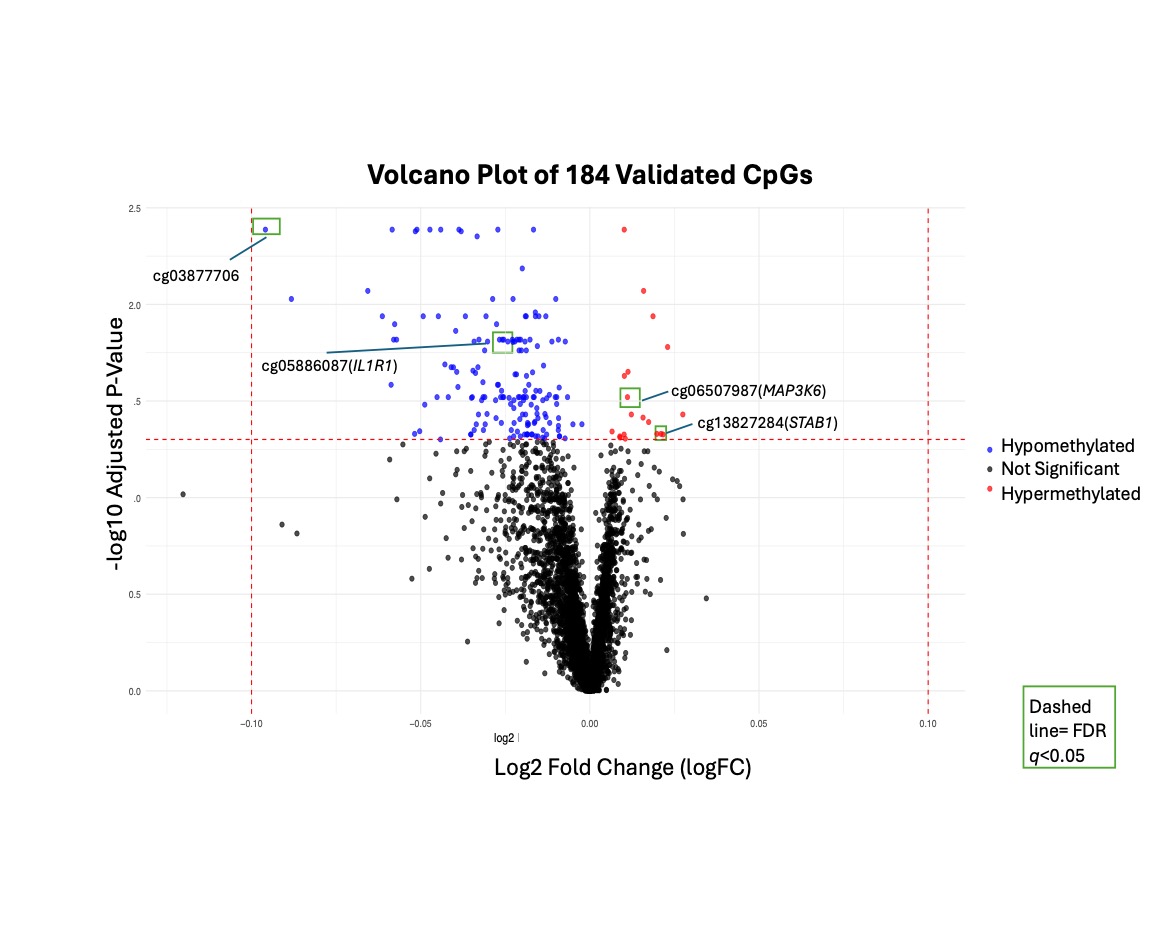 Figure 1. Volcano Plot of validated significant CpG sites.
Figure 1. Volcano Plot of validated significant CpG sites.
.jpg) Figure 2. Gene Set Enrichment Analysis (GSEA) with Gene Ontology.
Figure 2. Gene Set Enrichment Analysis (GSEA) with Gene Ontology.
To cite this abstract in AMA style:
Nelson M, Horton M, Nititham J, Yazdany J, Dall'Era M, Lanata C, Criswell L. DNA methylation patterns in systemic lupus erythematosus associated with nephritis status [abstract]. Arthritis Rheumatol. 2025; 77 (suppl 9). https://acrabstracts.org/abstract/dna-methylation-patterns-in-systemic-lupus-erythematosus-associated-with-nephritis-status/. Accessed .« Back to ACR Convergence 2025
ACR Meeting Abstracts - https://acrabstracts.org/abstract/dna-methylation-patterns-in-systemic-lupus-erythematosus-associated-with-nephritis-status/