Session Information
Session Type: ACR Abstract Session
Session Time: 11:00AM-12:30PM
Background/Purpose: Systemic lupus erythematosus (SLE) affects various organs and tissues, but whether pathologic processes in each organ are distinct or whether dysregulated molecular functions are found in common in all tissues is not known. We, therefore, conducted a meta-analysis of gene expression profiles in four affected SLE tissues to identify commonly dysregulated pathways.
Methods: Gene expression datasets for discoid lupus erythematosus (DLE), lupus arthritis (LA), lupus nephritis (LN) glomerulus (Glom), and LN tubulointerstitium (TI) were obtained from GEO. Differentially expressed genes (DEGs) were identified by LIMMA analysis for each dataset. DEGs from each tissue were analyzed with a multi-pronged bioinformatics approach to elucidate common immune cell infiltrates and common functional categories. These findings were then utilized to form modules of co-expressed genes to determine their enrichment using Gene Set Variation Analysis (GSVA).
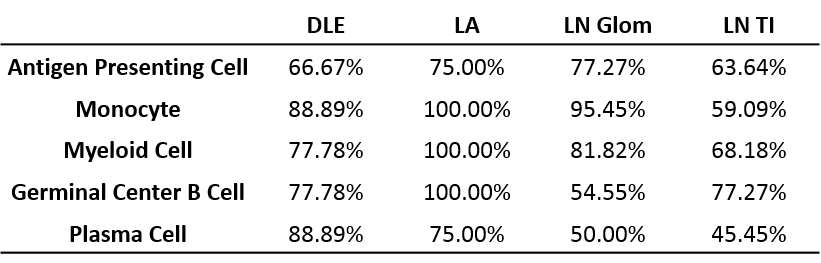
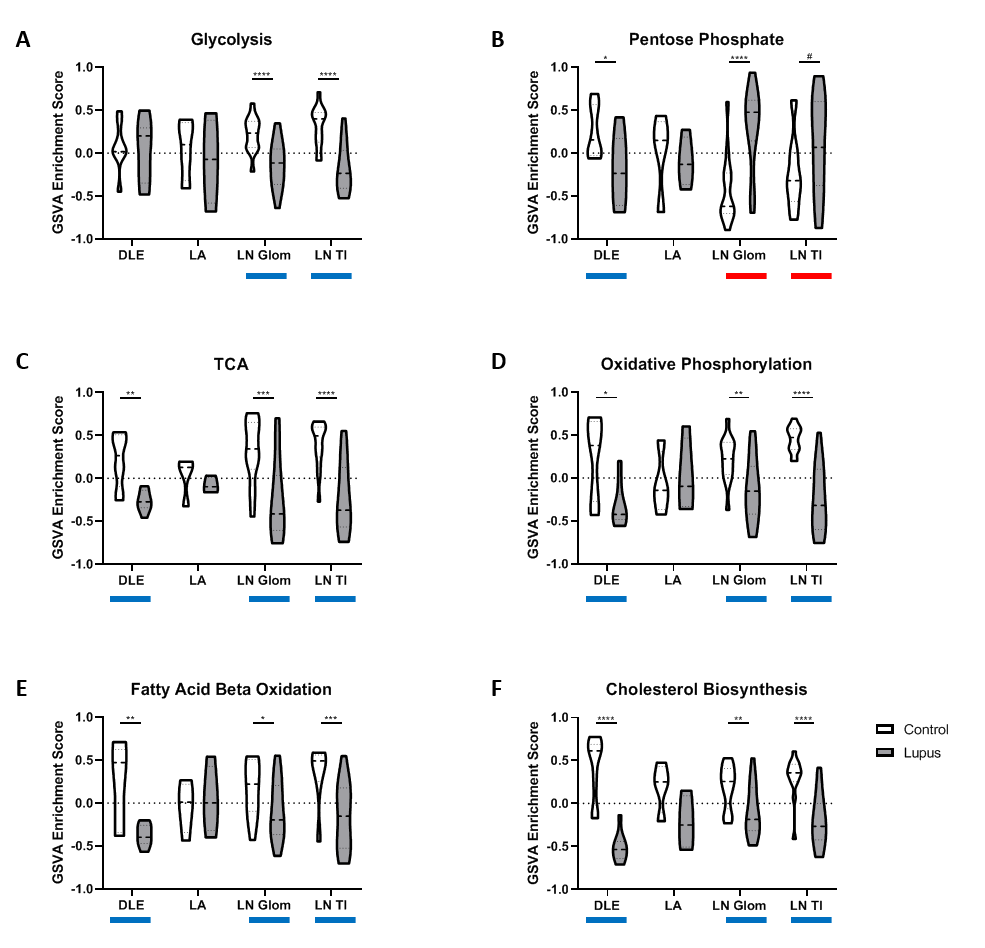
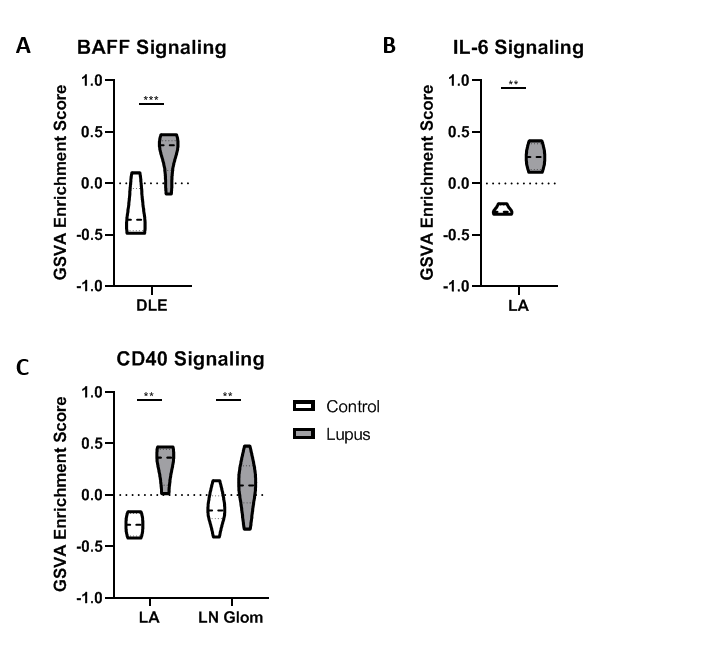
Results: All tissues demonstrated the presence of immune cells with the fewest immune cell transcripts in LN TI. Analysis of bulk gene expression revealed enrichment of antigen presenting cells (APCs), monocytes, and myeloid cells in all four tissues. Notably, enrichment of B cells, plasma cells, germinal center (GC) B cells, and CD8 T cells was only detected in DLE and LA. All four tissues demonstrated upregulated immune activity, including interferon-stimulated genes, pattern recognition receptors (PRRs), and antigen presentation (MHC Class II). Pro-apoptosis genes were also found enriched in DLE, LN Glom, and LN TI. A generalized decrease in biochemical processes was found in all four tissues and a specific decrease in both fatty acid biosynthesis and the tricarboxylic acid cycle was found in DLE and LN. Ingenuity Pathway Analysis (IPA®) further confirmed the activation of Dendritic Cell Maturation, Interferon, NFAT Regulation of Immune Response, PRRs, and TH1 signaling pathways in all four tissues. Additionally, IPA demonstrated cholesterol biosynthesis was decreased in all tissues except LA.
To confirm the aforementioned cellular infiltrates and aberrant pathways, as well as additional pathways, were operative in individual SLE tissues, GSVA was used to analyze enrichment of gene modules in patient samples. As shown in Table 1, Figure 1, and Figure 2, specific abnormalities were found in the majority of tissues, including enrichment of myeloid cells/monocytes, APCs, and GC B cells, whereas others were observed in some but not all tissues.
Conclusion: Common cellular infiltrates and molecular pathways were found in all affected tissues, suggesting commonalities in lupus organ pathogenesis. However, certain cell types and signaling were predominant in some tissues over others and GSVA illustrated heterogeneity between patients. Together this analysis informs a tissue-specific model of lupus immunopathogenesis and metabolic dysfunction with common and unique features and highlights the importance of patient specific identification of dysfunctional pathways in lupus organ pathogenesis.
To cite this abstract in AMA style:
Kingsmore K, Heuer S, Hubbard E, Catalina M, Bachali P, Lipsky P, Grammer A. Transcriptomic Meta-analysis of Lupus Affected Tissues Reveals Shared Immune, Metabolic, and Biochemical Dysregulation [abstract]. Arthritis Rheumatol. 2019; 71 (suppl 10). https://acrabstracts.org/abstract/transcriptomic-meta-analysis-of-lupus-affected-tissues-reveals-shared-immune-metabolic-and-biochemical-dysregulation/. Accessed .« Back to 2019 ACR/ARP Annual Meeting
ACR Meeting Abstracts - https://acrabstracts.org/abstract/transcriptomic-meta-analysis-of-lupus-affected-tissues-reveals-shared-immune-metabolic-and-biochemical-dysregulation/