Session Information
Session Type: Poster Session B
Session Time: 8:30AM-10:30AM
Background/Purpose: Systemic lupus erythematosus is a highly complex, heterogeneous, autoimmune disorder, with diverse clinical presentation and innate and adaptive immune system involvement. There is a need to better understand the molecular basis for dysregulated immune system function and disease pathophysiology in SLE in order to assign individuals to more homogeneous mechanistic subsets, enabling the identification of targets for more effective, cohort-specific, therapies with a higher impact in clinical improvement. Previous studies have used whole transcriptome data to identify key molecular features in order to achieve meaningful assignment of SLE patients into subsets defined to a large extent by predominate immune cell types in peripheral blood. These studies have allowed us to make inferences as to the contribution of specific immune cells to disease in different patient groups.
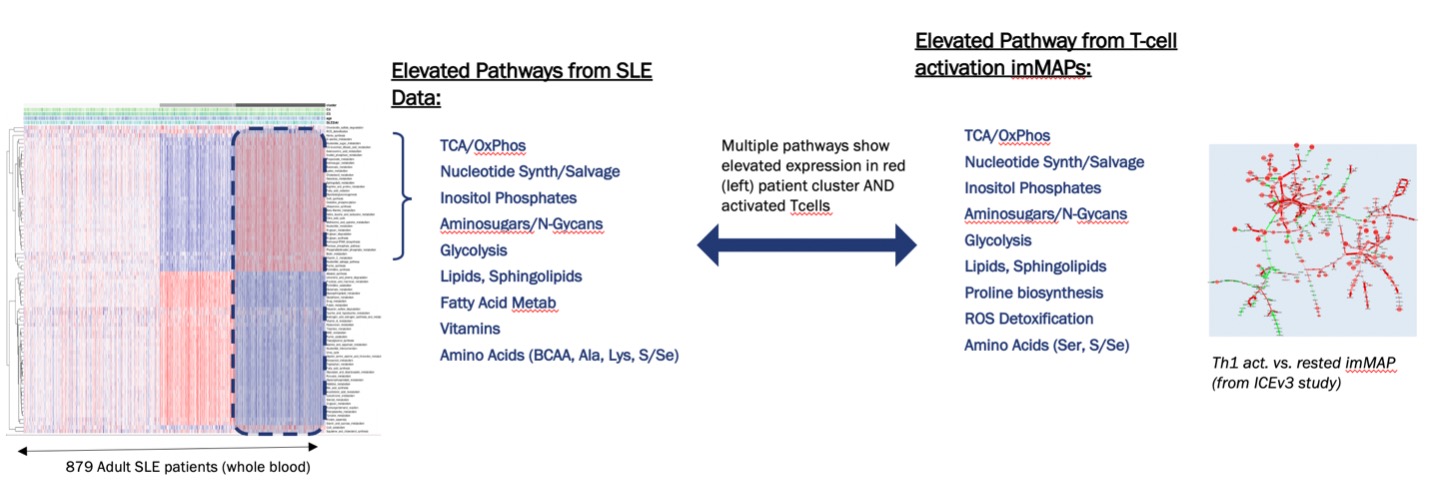
Methods: As metabolic regulation of innate and adaptive immune cells is increasingly appreciated as a driver of disease phenotypes, we sought to investigate if patient heterogeneity could be revealed by focusing solely on metabolic pathways relevant to the activation state of immune cells found in the peripheral blood. We collected a set of 84 metabolic pathways and gene sets, comprised of a total of 1,692 genes, and calculated the pathway eigengenes as described elsewhere (1). We applied our methodology to the ILLUMINATE-1 clinical trial cohort transcriptomics data set (2), generated from whole blood samples collected from 879 patients. After calculating the pathway eigengenes for the patient cohort, we performed unsupervised clustering using 30 different algorithms, in order to determine the optimal number of clusters in these data (Fig 1.) Additionally, we calculated the predicted immune cell population for each patient as described elsewhere (3), enabling us to ascribe biological significance to the identified clusters. Our results show that, when comparing the patient clusters on whole transcriptome data versus on pathway eigengenes, there is an enrichment in the immune cell populations in the latter (Fig 2), reflecting not only their proportion but also their activation or effector state. Furthermore, we compared the clustering results based on the pathway eigengenes with our immunometabolism map (imMAP), an algorithm for the integration of transcriptional and metabolic profiles, run on data from primary human effector memory peripheral blood T cells activated by anti-CD3/CD28/CD2, highlighting how the high lymphocytic cluster preserves features captured by our imMAP (Fig 3.)
Results: Our results indicate that SLE patients can be differentiated based upon metabolic features with potential important biological and clinical significance, given the increasing appreciation for disease-associated immune cell metabolic reprogramming in autoimmune diseases (4, 5).
Conclusion: Insights from these studies will facilitate discovery of novel targets that impact the disease-associated metabolic programming of immune cell drivers of SLE and provide a framework for precision medicine approaches to assign individual patients to disease subgroups that would best benefit from drugs modulating those targets.


To cite this abstract in AMA style:
Camacho D, Swantek J, Soh K, Kamphorst J, Kaimal V, Monroe J, Driggers E. Transcriptional Subsetting of SLE Patient Cohorts Based on Metabolic Pathway Activity [abstract]. Arthritis Rheumatol. 2021; 73 (suppl 9). https://acrabstracts.org/abstract/transcriptional-subsetting-of-sle-patient-cohorts-based-on-metabolic-pathway-activity/. Accessed .« Back to ACR Convergence 2021
ACR Meeting Abstracts - https://acrabstracts.org/abstract/transcriptional-subsetting-of-sle-patient-cohorts-based-on-metabolic-pathway-activity/