Session Information
Date: Monday, October 27, 2025
Title: (1147–1190) Miscellaneous Rheumatic & Inflammatory Diseases Poster II
Session Type: Poster Session B
Session Time: 10:30AM-12:30PM
Background/Purpose: Inflammatory lung disease (ILD) in Still’s disease (SD) has recently been described. Pulmonary arterial hypertension (PAH), a rare subtype of pulmonary hypertension (PH), is a potentially life-threating complication of SD. It is crucial to differentiate PAH (group 1 PH), characterized by progressive remodeling of small pulmonary arteries, from PH associated with interstitial lung disease (PH-ILD), which falls under group 3 PH and results primarily from parenchymal lung involvement and hypoxic vasoconstriction. Although in the context of SD both mechanisms, PH-ILD and PAH, may be involved, PAH has rarely been reported. This work reports a large cohort of PAH in adults with SD.
Methods: We identified 16 adult SD patients with PAH (PAH+ group, PAH was confirmed by right heart catheterization in all patients) by a call for observations from the CRIIMIDIATE network and a search of the French PH network registry. Patient characteristics and disease evolution were retrospectively compared with those of 111 SD controls without PAH (PAH-) followed in a reference center.
Results: The profile of patients in the PAH+ and PAH- groups differed (Table): 100% versus 69.4% female (p=0.006), 75% versus 17.1% Black (p< 0.0001), more active SD both at diagnosis and throughout the disease course, and more likely to present MAS (62.5% vs 14.4%, p< 0.0001), which was recurrent (i.e. ≥ 2 episodes) in most cases (7/10 patients), and exhibit eosinophilia during the disease course (68.7% vs 7.2%, p< 0.0001). Twelve patients in the PAH+ group presented mild radiological parenchymal features on the CT scan (interlobular septal thickening, non-specific ground-glass opacities) that could not explain PH. For the 84/127 patients with genetic typing, the HLA-DRB1*15 allele was more prevalent in PAH+ than PAH- patients (8/11 [72.7%] vs 22/51 [30.1%], p=0.014). The groups did not differ in treatment, except for methotrexate (81.2% vs 50.4%, p=0.029), canakinumab (50% vs 21.6%, p=0.026) or immunosuppressant agents (56.2% vs 10.8%, p< 0.0001) that were more often used in the PAH+ group, reflecting a more active underlying SD. There was higher frequency of drug reactions to interleukin 1 (IL-1) and/or IL-6 inhibitors in PAH+ than PAH- patients (37.5% vs 7.2%, p=0.002). Mortality was higher in PAH+ than PAH- patients (37.5% vs 0.9%, p< 0.0001), all deaths related to SD flare (Figure).
Conclusion: PAH develops on the same background as other manifestations of SD-related ILD (i.e., in patients with MAS, especially when recurrent; eosinophilia; and carrying the HLA-DRB1*15 allele). Female sex and Black ethnicity seem to be risk factors. This work reinforces the association of the HLA-DRB1*15 allele with severe forms of SD and raises the question of treatment optimizations to better control both SD and its complications. The fact that PAH can be isolated in SD, i.e., it can occur in the absence of major parenchymal involvement, should encourage physicians to screen for it using echocardiography in cases of unexplained dyspnoea, before confirmation by catheterization. The existence of PAH has major therapeutic implications, since it requires the addition of vasodilating agents to targeted and non-targeted immunomodulating therapies.
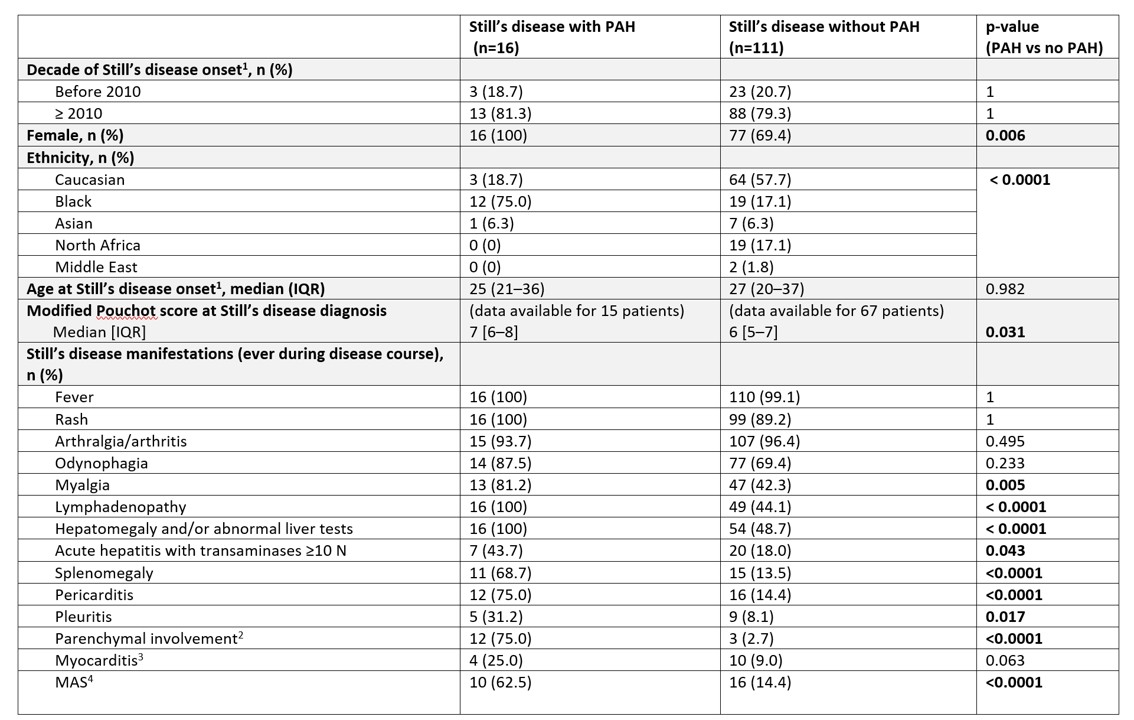 Table 1A. Characteristics of Still’s disease patients with and without pulmonary arterial hypertension (PAH)
Table 1A. Characteristics of Still’s disease patients with and without pulmonary arterial hypertension (PAH)
1In some patients, the disease developed in childhood (systemic juvenile idiopathic arthritis), but symptoms persisted and/or recurred in adulthood.
2The radiological parenchymal involvement was mild in the 12 patients of the PAH+ group: interlobular septal thickening in all cases, ground-glass opacities of non-specific nature in half of the cases (please see main text and figure 1 for details). The radiological parenchymal involvement was mild to moderate on the CT scan for the 3 patients of the PAH- group who had parenchymal involvement (please see supplementary tables 16 and 17 for details). Importantly, no patient in either group presented with pulmonary alveolar proteinosis and endogenous lipoid pneumonia.
3Including one patient with PAH in whom myocarditis developed in the context of major eosinophilia up to 8,000/mm3.
4Among the population with PAH, 7 patients had multiple (≥2) episodes of MAS, and 1 had a refractory episode of MAS over several months associated with uncontrolled PAH that led to death.
csDMARDs, conventional synthetic disease-modifying anti-rheumatic drugs–IL, interleukin–IQR, interquartile range–JAK, Janus kinase–MAS, macrophage activation syndrome–PAH, pulmonary arterial hypertension–10N, 10 times above normal
.jpg) Table 1B (table continued). Characteristics of Still’s disease patients with and without pulmonary arterial hypertension (PAH)
Table 1B (table continued). Characteristics of Still’s disease patients with and without pulmonary arterial hypertension (PAH)
5Eosinophilia was defined as absolute eosinophil count ≥ 700/mm3 or eosinophils ≥10% of the total white blood cell count, measured on at least two occasions (Lerman et al, Arthritis Care & Research. 2023).
6Some patients had several lines of treatment.
7Drug reactions to IL-1 and/or IL-6 inhibitors were all confirmed by a dermatologist and/or an allergologist, and included rashes atypical for SD (e.g. nonevanescent, pruritic, urticarial, a fortiori in the presence of blood eosinophilia), or acute reactions (respiratory distress, swelling or Quincke edema during or near the time of medication administration). Two patients in the HTAP+ group met the criteria for DRESS syndrome (RegiSCAR for DRESS ≥ 4) (Kardaun et al, Br J Dermatol. 2013). Local reactions at the injection site were not included in this category, as they are common with subcutaneous biotherapies.
csDMARDs, conventional synthetic disease-modifying anti-rheumatic drugs–IL, interleukin–IQR, interquartile range–JAK, Janus kinase–MAS, macrophage activation syndrome–PAH, pulmonary arterial hypertension–10N, 10 times above normal
.jpg) Figure. Overall survival after pulmonary arterial hypertension diagnosis in patients with Still’s disease (n=16).
Figure. Overall survival after pulmonary arterial hypertension diagnosis in patients with Still’s disease (n=16).
Overall survival at 1 and 3 years was 81% and 75%, respectively.
To cite this abstract in AMA style:
Mitrovic S, Boucly A, Carmagnat M, Savale L, Jaïs X, Taupin J, Lazaro E, Berthoux E, schleinitz n, Ghigna M, joanna k, Mariette X, Roussin C, Juge P, Humbert M, Sitbon O, Montani D, Fautrel B. Pulmonary Arterial Hypertension in Adults with Still’s Disease: Another Pulmonary Manifestation Associated with HLA-DRB1*15 [abstract]. Arthritis Rheumatol. 2025; 77 (suppl 9). https://acrabstracts.org/abstract/pulmonary-arterial-hypertension-in-adults-with-stills-disease-another-pulmonary-manifestation-associated-with-hla-drb115/. Accessed .« Back to ACR Convergence 2025
ACR Meeting Abstracts - https://acrabstracts.org/abstract/pulmonary-arterial-hypertension-in-adults-with-stills-disease-another-pulmonary-manifestation-associated-with-hla-drb115/