Session Information
Session Type: Abstract Session
Session Time: 2:00PM-3:30PM
Background/Purpose: Genome-wide association studies in systemic lupus erythematosus (SLE) have linked loss-of-function mutations in phagocytic NADPH oxidase complex (NOX2) genes, including NCF1 and NCF2, to disease pathogenesis. The prevailing model holds that reduced NOX2 activity promotes SLE via defective efferocytosis, the immunologically silent clearance of apoptotic cells. However, we previously showed that B cell-intrinsic Toll-like receptor (TLR) signaling is modulated by endolysosomal trafficking, such that dysregulated endosomal flux can drive breaks in B cell tolerance. Since NCF1/NCF2 are known regulators of endosomal trafficking in myeloid lineages, we hypothesized that a parallel B cell-intrinsic mechanism contributes to lupus risk.
Methods: We tested the impact of NOX2 family gene deletion on B cell TLR signaling using in vivo animal models, primary murine B cells, NCF1-null human B cell lymphoma lines, and CRISPR-edited primary human B cells
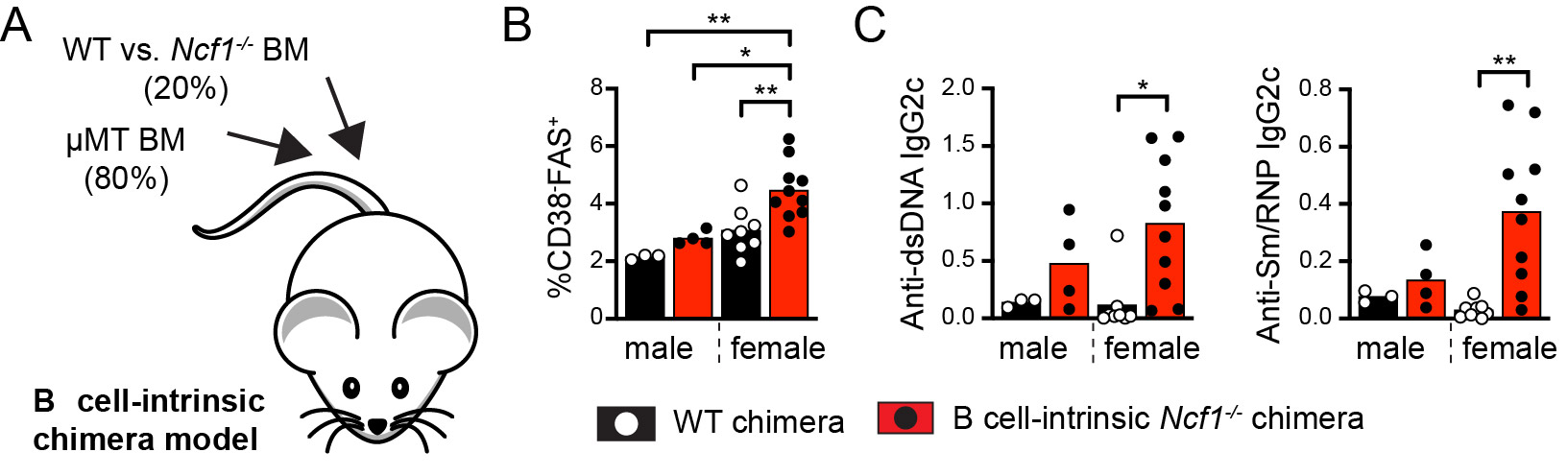
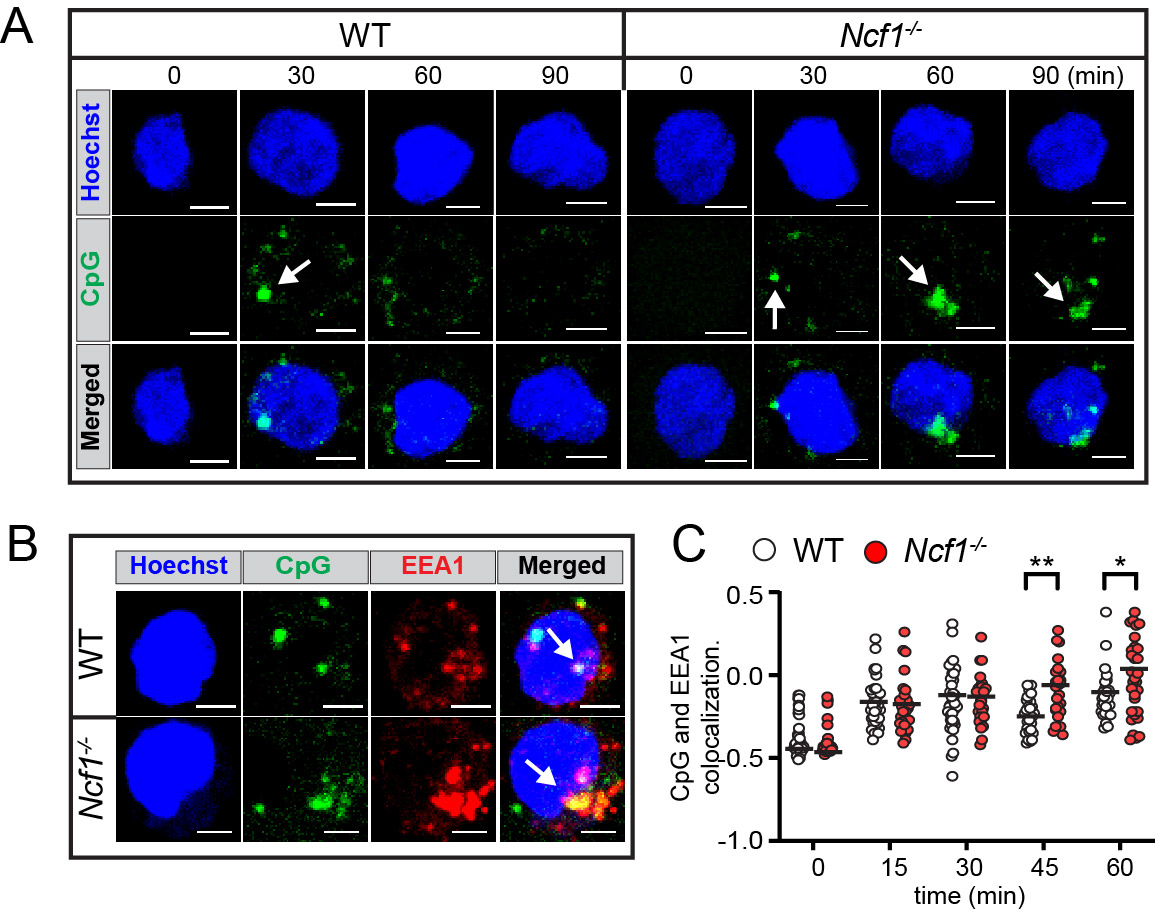
Results: We show that NOX2-deficient mice exhibited increased humoral responses to nucleic acid-containing antigens, findings which correlate with enhanced B cell signals downstream of endosomal TLRs. In keeping with important roles for B cell TLRs in lupus pathogenesis, B cell-intrinsic NADPH oxidase deletion promoted murine humoral autoimmunity (Figure 1). To understand the underlying mechanisms, we quantified TLR-induced intracellular trafficking in B cells using live cell microscopy. Following CpG stimulation, NADPH oxidase activation facilitated trafficking of TLR-containing endosomes to lysosomes, resulting in TLR signal termination. Whereas initial uptake and aggregation of fluorescent CpG in early endosomes was preserved in NCF1-null B cell, loss of NADPH oxidase activity limited signal degradation resulting in enhanced downstream NFkB activation (Figure 2). Finally, CRISPR-mediated disruption of NCF1 in primary human B cells confirmed a direct role for NOX2 in regulating endosomal TLR signaling in B cells. In response to in vitro TLR9 activation, NCF1-deficient human B cells exhibited increased differentiation into IgM- and IgG-producing plasma cells, supporting a mechanistic link between B cell NADPH oxidase activity and human lupus pathogenesis.
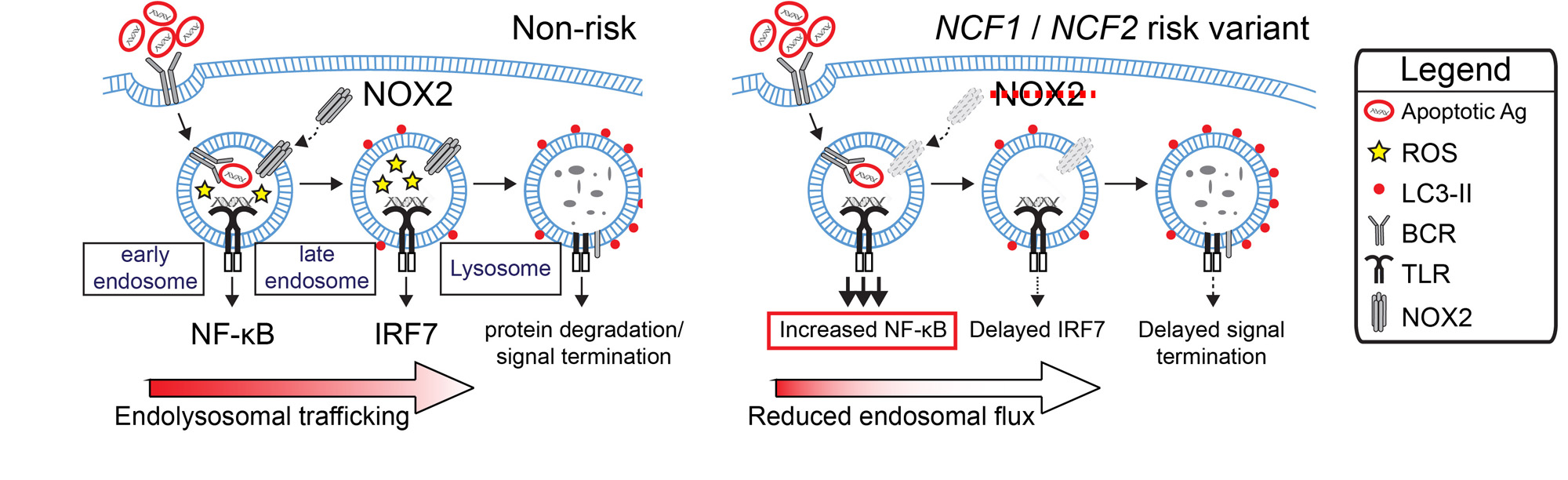
Conclusion: Together, these data highlight a new B cell-specific mechanism contributing to autoimmune risk in NCF1 and NCF2variant carriers. We show that loss of NADPH oxidase results in disruption of B cell endolysosomal trafficking, thereby lowering endosomal TLR signaling thresholds and promoting B cell-intrinsic breaks in tolerance (disease model shown in Figure 3).
To cite this abstract in AMA style:
Liu S, Shumlak N, Largent A, Lewis S, Acharya M, Jackson S. NADPH Oxidase Exerts a B Cell-intrinsic Contribution to Lupus Risk by Modulating Endosomal Toll-like Receptor (TLR) Signals [abstract]. Arthritis Rheumatol. 2023; 75 (suppl 9). https://acrabstracts.org/abstract/nadph-oxidase-exerts-a-b-cell-intrinsic-contribution-to-lupus-risk-by-modulating-endosomal-toll-like-receptor-tlr-signals/. Accessed .« Back to ACR Convergence 2023
ACR Meeting Abstracts - https://acrabstracts.org/abstract/nadph-oxidase-exerts-a-b-cell-intrinsic-contribution-to-lupus-risk-by-modulating-endosomal-toll-like-receptor-tlr-signals/