Session Information
Date: Wednesday, November 16, 2016
Title: Systemic Sclerosis, Fibrosing Syndromes and Raynaud's – Pathogenesis, Animal Models and Genetics II
Session Type: ACR Concurrent Abstract Session
Session Time: 9:00AM-10:30AM
Background/Purpose: Clinical trials in SSc have tended to be underpowered and not meet clinical endpoints. Genome-wide gene expression measured in some studies can prove challenging to analyze and interpret due to the small sample sizes. Often, few differentially expressed genes can be detected pre- and post-treatment after multiple hypothesis testing correction. Here, we have leveraged data from thousands of experiments and utilized state-of-the-art machine learning to robustly identify genes and processes that are predicted to be modulated during clinically significant response to treatment and view the processes modulated in each through the lens of a functional genomic network.
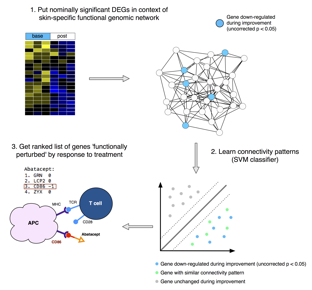
Methods: We analyzed immunomodulatory therapeutics: abatacept (Chakravarty et al., 2015) (CTLA4-IgG), MMF (Hinchcliff et al., 2013; Mahoney et al., 2015), and rituximab (Lafyatis et al., 2009; Pendergrass et al., 2012) (anti-CD20), as well as a tyrosine kinase inhibitor (TKI): nilotinib (Gordon et al., 2015), and a monoclonal antibody to the pro-fibrotic cytokine TGF-β: fresolimumab (Rice et al., 2015). We applied a common improvement criterion to each study: -20% OR -5 modified Rodnan Skin Score (mRSS) (Khanna et al., 2006). Nominally significant genes that changed in each trial were placed in the context of a skin-specific functional genomic network and their connectivity patterns learned using a support vector machine (SVM) (Greene et al., 2015) (Figure 1).
Results: Our machine learning approach captures features beyond differential expression and is better at identifying predicted gene targets of each therapy than the t-statistic alone. As an example, CD86 is captured when abatacept is analyzed due to its connectivity in the network, although the gene itself is not differentially expressed. We observe the abrogation of inflammatory pathways in improvers in multiple studies regardless of the mechanism of action of a drug, which suggests high expression of immune-related genes may represent an active disease state. The framework allows us to compare different trials and ask if the patients that failed one therapy could possibly improve on a different therapy. As an example, we find that genes with high expression at baseline in fresolimumab non-improvers were downregulated in MMF improvers, suggesting that these patients might have benefited from the combination therapy.
Conclusion: This work provides a systems biology framework for analysis of therapeutic trials in rare diseases and for prediction of potentially beneficial treatment combinations. Our results suggest that if a strong inflammatory signature is present in a patient it may need to be treated with an immunosuppressive treatment, but that this may show more efficacy if paired with an anti-fibrotic drug such as anti-TGF-β therapy, especially for patients with a weak baseline inflammatory signature.
To cite this abstract in AMA style:
Taroni JN, Martyanov V, Whitfield ML. Meta-Analysis of SSc Clinical Trials with Molecular Gene Expression Data Suggests Potential Combination Therapies [abstract]. Arthritis Rheumatol. 2016; 68 (suppl 10). https://acrabstracts.org/abstract/meta-analysis-of-ssc-clinical-trials-with-molecular-gene-expression-data-suggests-potential-combination-therapies/. Accessed .« Back to 2016 ACR/ARHP Annual Meeting
ACR Meeting Abstracts - https://acrabstracts.org/abstract/meta-analysis-of-ssc-clinical-trials-with-molecular-gene-expression-data-suggests-potential-combination-therapies/