Session Information
Session Type: Abstract Submissions (ACR)
Background/Purpose: Osteoarthritis (OA) is a prevalent joint disease, with several identified clinical risk factors. However, the search for genetic risk factors by candidate gene and genome-wide association studies of human OA populations has identified only a small number of candidate genes and pathways potentially involved in the disease. Thus, we aim to build a genome-wide molecular profile of OA to elucidate regulatory mechanisms of OA pathogenesis and to identify possible therapeutic targets.
Methods: We extracted total RNA from full-thickness articular cartilage from 18 human donors (8 normal and 10 OA) and performed mRNA and microRNA sequencing on an Illumina HiSeq 2000 (single-end, 100bp reads). DNA was extracted from full-thickness articular cartilage from 23 different human donors (11 normal and 12 OA) and we performed a DNA methylation assay on the Illumina Infinium HumanMethylation450 BeadChip. The RNA sequencing data were analyzed using the Tuxedo package to align raw reads to the human genome, quantify gene expression and perform differential expression analysis. We used the Bioconductor package ChAMP to perform differential methylation analysis. We employed downstream functional enrichment analyses for transcription factors and microRNAs using Fisher’s Exact Test, pathway analyses using the Bioconductor package SPIA, and network analyses using Cytoscape. Finally, we created an interaction network using the Human Integrated Protein-Protein Interaction Reference using enriched and additional OA related transcription factors. This network was annotated with differentially expressed mRNAs, microRNAs with validated targets in this network and their methylation status.
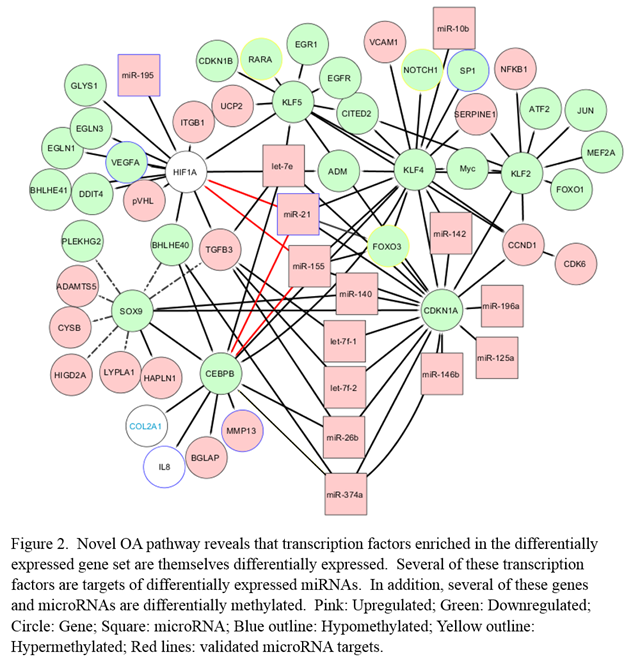
Results: Several genes, microRNAs and transcription factors are differentially expressed and reveal unique molecular signatures of OA (Figure 1). Many of these genes and microRNAs possess differentially methylated promoters. In addition, they are key regulators of a novel OA regulatory network (e.g., miR-21, miR-155, CEBPB, FOXO3) (Figure 2). We validated the microRNA targets in this network and the hypomethylation of the miR-21 promoter. We discovered that miR-21 targets the transcription factors HIF1A and CEBPB, providing a regulatory mechanism for the downregulation of these transcription factors and their targets.
Conclusion: Our findings reveal a complex regulatory interaction network of OA pathogenesis controlled by microRNAs, transcription factors and DNA methylation and suggest therapeutic targets to treat or delay disease progression.

Disclosure:
K. M. Fisch,
None;
R. Akagi,
None;
O. Alvarez-Garcia,
None;
T. Teramura,
None;
Y. Muramatsu,
None;
M. Saito,
None;
S. Duffy,
None;
S. Grogan,
None;
T. Sasho,
None;
D. D’Lima,
None;
A. I. Su,
None;
M. K. Lotz,
None.
« Back to 2014 ACR/ARHP Annual Meeting
ACR Meeting Abstracts - https://acrabstracts.org/abstract/integrative-omics-profiling-reveals-dysregulated-novel-pathways-mediated-by-micrornas-and-dna-methylation-in-osteoarthritis/