Session Information
Session Type: Poster Session B
Session Time: 10:30AM-12:30PM
Background/Purpose: Pulmonary arterial hypertension (PAH) is a severe and frequently life-threatening complication of systemic sclerosis (SSc). Treatment of SSc-PAH follows the same approach of idiopathic PAH and mainly relies on the combined action of vasodilators and anti-proliferative agents. Whilst current treatment guidelines suggest that immunosuppression (IS) improves outcome of PAH in some connective tissue diseases (i.e. mixed connective tissue disease; MCTD, or systemic lupus erythematosus; SLE), it is not recommended for SSc-PAH. The aim of this study was to retrospectively ask whether early IS could impact frequency or timing of development of PAH in patients with SSc, and influence survival in SSc-PAH.
Methods: In this retrospective study we included patients meeting the 2013 ACR/EULAR classification criteria for SSc, followed up at our centre from 1998 to 2024. Early IS was defined as treatment in the first 5 years since SSc diagnosis with either conventional synthetic DMARD (mycophenolate mofetil, azathioprine, cyclosporine, hydroxychloroquine, cyclophosphamide), prednisolone equivalent dose≥10mg daily, or rituximab. Being this an historical cohort, pre-capillary pulmonary hypertension (precap-PH) was defined using the 2015 ESC/ERS criteria (mPAP ≥ 25mmHg, PVR >3 WU, PWP ≤15mmHg). PAH was defined as precap-PH in absence of extensive interstital lung disease (i.e. ≥20% at high resolution CT scan).
Outcomes of interest were development of PAH, time from SSc diagnosis to PAH diagnosis, and survival in patients with PAH. Descriptive statistics, logistic and linear regression, and survival analysis were performed with STATA-18 software package, using significant variables as confounders.
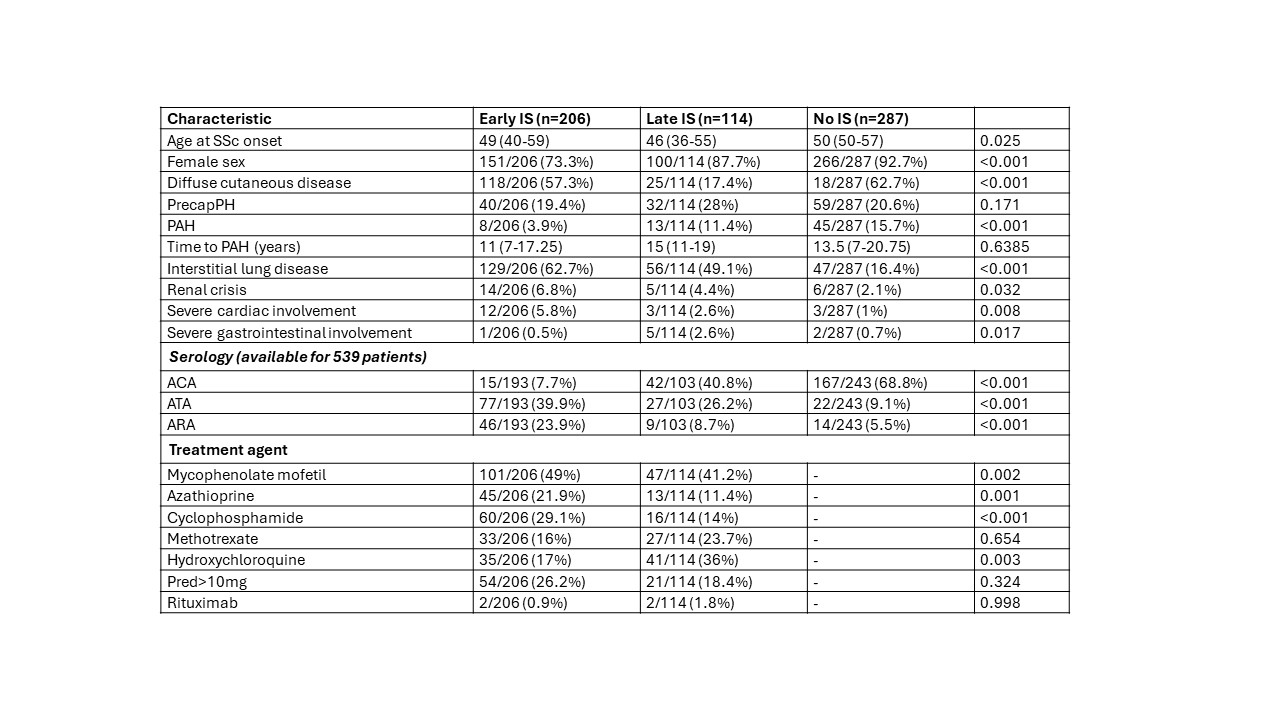
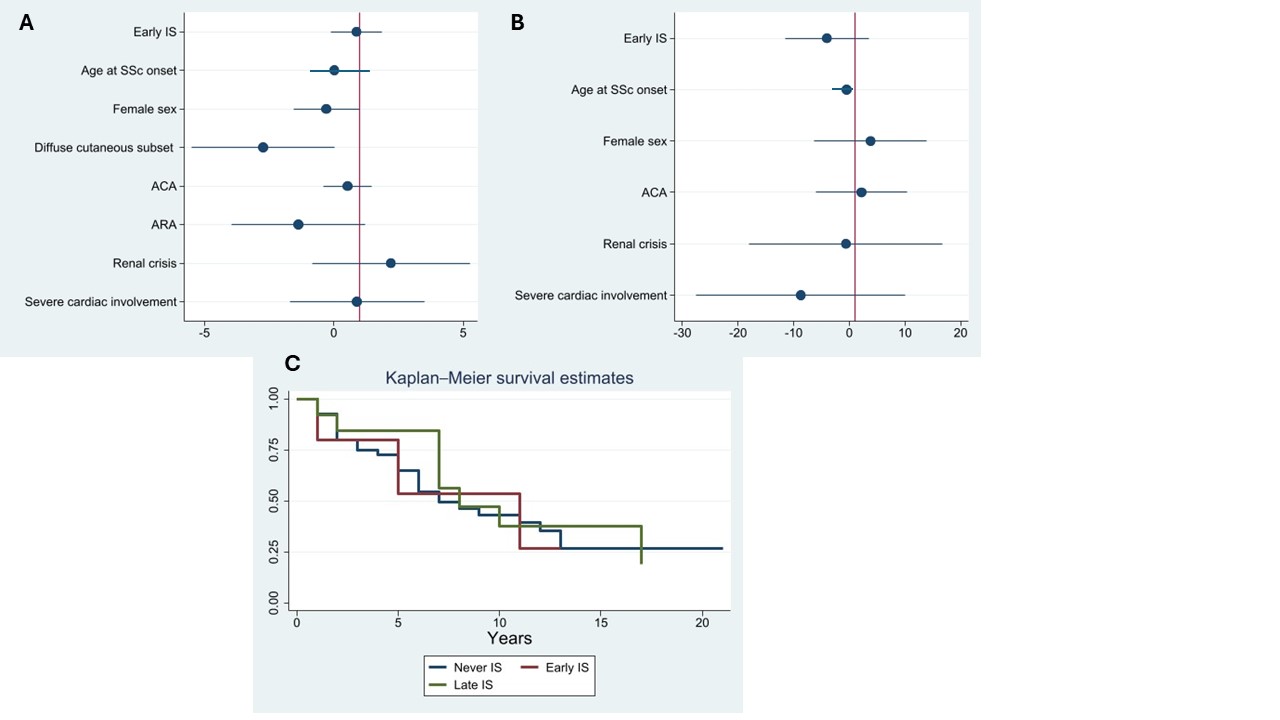
Results: We evaluated 629 patients from our electronic records. Complete clinical and therapy information were available for 607 patients, who were included in the analysis (Table 1). Two-hundred and six patients received early IS: they were more commonly male, with diffuse cutaneous subset, frequent internal organ involvement and high rates of positivity for anti-topoisomerase I antibody (ATA) and anti-RNA polymerase III antibody (ARA), with lower rates for anti-centromere antibody (ACA). Early IS was associated with a protective effect on PAH development (OR 0.24, C.I. 0.11 – 0.52, p< 0.0001). However, the protective effect is lost when adjusting for major confounders (sex, diffuse cutaneous subset, internal organ involvement, serology). Similarly, time from diagnosis of SSc to development of PAH was not impacted by early IS (Figure 1). Considering only patients with PAH, survival analysis (Figure 1) demonstrated similar survival curves between early IS, late IS and patient never subjected to IS.
Conclusion: Despite overall differences in frequency between groups, our analysis suggests IS, whether early or late, does not impact on development and survival in SSc-PAH once other relevant disease characteristics are considered. Our findings, although limited by the retrospective approach, confirm that the pathogenesis of SSc-PAH is likely aligned with idiopathic PAH, with little contribution of immune-inflammation, unlike SLE or MCTD.
To cite this abstract in AMA style:
Rodolfi S, Ng C, Ruiz Bejerano A, Kanitkar M, Ong V, Denton C. Impact of Immunosuppressive Treatment on Development and Survival in Systemic Sclerosis Associated Pulmonary Arterial Hypertension (SSc-PAH) [abstract]. Arthritis Rheumatol. 2024; 76 (suppl 9). https://acrabstracts.org/abstract/impact-of-immunosuppressive-treatment-on-development-and-survival-in-systemic-sclerosis-associated-pulmonary-arterial-hypertension-ssc-pah/. Accessed .« Back to ACR Convergence 2024
ACR Meeting Abstracts - https://acrabstracts.org/abstract/impact-of-immunosuppressive-treatment-on-development-and-survival-in-systemic-sclerosis-associated-pulmonary-arterial-hypertension-ssc-pah/