Session Information
Session Type: Poster Session D
Session Time: 1:00PM-3:00PM
Background/Purpose: Diffuse alveolar hemorrhage (DAH) is a life-threatening syndrome with bleeding from the pulmonary microvasculature. It is classified as pulmonary capillaritis if histology shows neutrophilic invasion with fibrinoid necrosis of capillary walls. This can occur secondary to systemic autoimmune disease or in isolation as idiopathic pulmonary capillaritis (IPC). There is a paucity of literature describing IPC in pediatrics. We describe the clinical presentation, management, and outcomes of a pediatric cohort with biopsy-proven IPC compared to other etiologies of DAH, including idiopathic pulmonary hemosiderosis (IPH) and autoimmune disease (DAH-AD).
Methods: ICD-10 codes were used to identify patients 1-18 years of age who presented to Texas Children’s Hospital from 2011-2021 with concerns for pulmonary hemorrhage. Inclusion criteria were radiographic evidence of bilateral hemorrhage, anemia, and clinical respiratory insufficiency in patients. Exclusion criteria were anticoagulation use, perioperative period, and antecedent trauma, including CPR. IPC patients had capillaritis on biopsy and no evidence of systemic autoimmune disease at presentation. IPH patients had no vasculitis or capillaritis on biopsy and no evidence of systemic autoimmune disease. We extracted clinical features, diagnostics, immunomodulation use, and outcomes.
Results: 680 patient charts included an ICD-10 code suggestive of pulmonary hemorrhage. 192 patients met inclusion and exclusion criteria for DAH. Of these, 10 met criteria for IPC, 7 for IPH, and 54 for DAH-AD with 24/54 (44%) ANCA vasculitis and 18/54 (33%) lupus. Majority (65%) of DAH-AD patients presented with hemorrhage at time of systemic diagnosis. One additional IPC patient was later reclassified as COPA syndrome, and one additional IPH patient, who had no vasculitis on original biopsy, later developed pANCA and MPO antibodies with recurrent DAH.
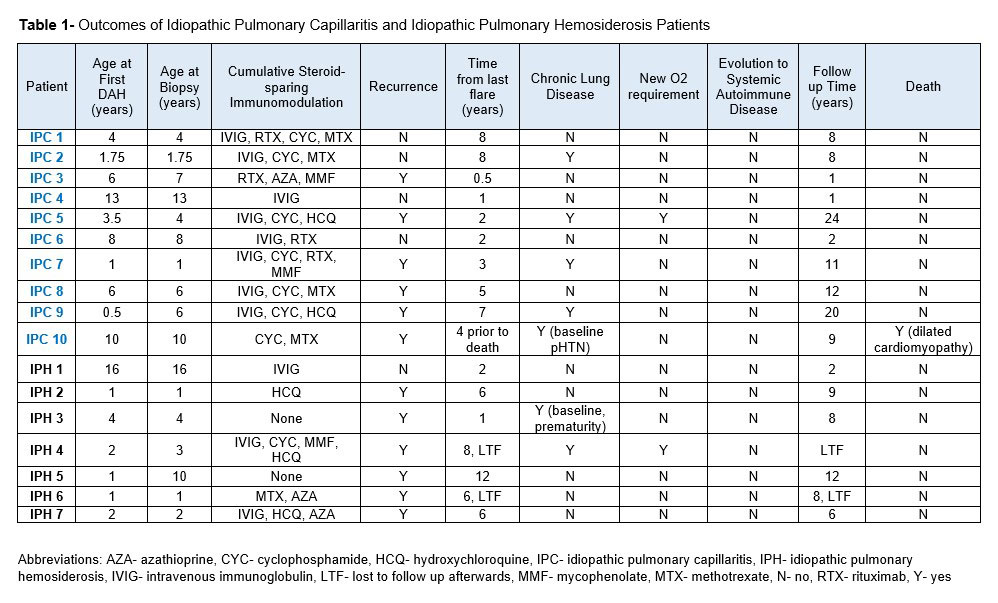
Median age of the IPC patients at first DAH presentation was 5 years (range 0.5-13). 5 were evaluated for symptomatic anemia prior to presentation, and 5 reported hemoptysis at presentation. All received high-dose IV corticosteroids, and 8 received IVIG. 9 patients received additional immunomodulation: 5 cyclophosphamide, 2 rituximab, and 2 both agents. 8 received steroid-sparing maintenance therapy with azathioprine, hydroxychloroquine, IVIG, methotrexate, or mycophenolate. Despite these interventions, 6 IPC patients had a refractory course with recurrent DAH. This is in comparison to 6/7 (86%) with IPH and 14/54 (26%) with DAH-AD. One IPC patient developed severe interstitial lung disease with baseline oxygen requirement and is being evaluated for lung transplant 24 years after initial DAH. One patient with IPC died, secondary to comorbidity (dilated cardiomyopathy). Table 1 outlines IPC and IPH outcomes.
Conclusion: Although treated aggressively, children with biopsy-proven IPC at our institution had a more chronic course with more recurrent DAH as compared to IPH and DAH-AD patients. The majority of IPC and IPH patients did not develop systemic autoimmune disorders. Additional prospective cohort studies are needed to determine predictors of recurrence and optimal treatment course of DAH phenotypes.
To cite this abstract in AMA style:
Chun A, vogel T, Ramirez A, De Guzman M, Muscal E, Silva-Carmona M. Idiopathic Pulmonary Capillaritis Within the Spectrum of Pediatric Diffuse Alveolar Hemorrhage [abstract]. Arthritis Rheumatol. 2022; 74 (suppl 9). https://acrabstracts.org/abstract/idiopathic-pulmonary-capillaritis-within-the-spectrum-of-pediatric-diffuse-alveolar-hemorrhage/. Accessed .« Back to ACR Convergence 2022
ACR Meeting Abstracts - https://acrabstracts.org/abstract/idiopathic-pulmonary-capillaritis-within-the-spectrum-of-pediatric-diffuse-alveolar-hemorrhage/