Session Information
Date: Monday, October 22, 2018
Title: Systemic Sclerosis and Related Disorders – Basic Science Poster II
Session Type: ACR Poster Session B
Session Time: 9:00AM-11:00AM
Systemic sclerosis (SSc) is a heterogeneous autoimmune disorder with poor outcomes and no FDA-approved therapies. Prior work has shown gene expression patterns associated with inflammation and innate immune system activation in multiple affected organ systems. Networks of activating transcription factors (TF) and miRNAs that underlie SSc molecular subsets and clinical co-variates have not been characterized in detail. We analyzed transcriptional regulatory networks underlying the inflammatory intrinsic gene expression subset of SSc and associated clinical covariates.
Methods:
Gene expression profiles were downloaded from Milano et al (GSE9285; 75 samples), Pendergrass et al (GSE32413; 89 samples), Hinchcliff et al (GSE59787; 165 samples) and Assassi et al (GSE58095; 102 samples). In total 431 samples from 244 patients were analyzed. The C3 target gene set (836 regulators) was downloaded from the MSigDB. Gene expression and target gene profiles were integrated using the BASE algorithm to calculate sample-specific regulator activity scores. TF activity scores were positively correlated and miRNAs activity scores were negatively correlated with gene expression. Correlations were calculated between activity scores and samples MRSS. Regulators were selected from those that had correlations greater than 0.15 or lesser than -0.15 in at least 3 cohorts. A transcriptional interaction network was created by Cytoscape.
Results:
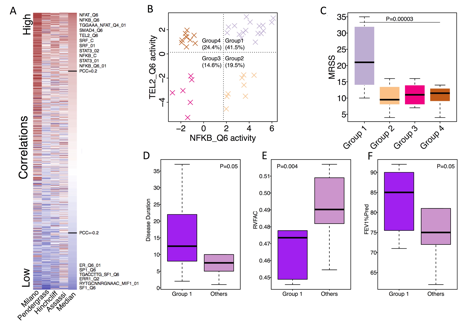
We identified 60 regulators whose activity scores were consistently, positively correlated with MRSS in inflammatory patients in all datasets. Examples include core binding factors, innate immune-related pathways, and telomere maintenance (NFAT, NFKB, STAT3, TEL2; Fig. 1A). We also found that using the activities of NFKB and TEL2, inflammatory samples were clustered into 4 subtypes (Fig. 1B). ÔDouble highÕ samples in group 1 had much worse skin disease (Fig. 1C, P=0.00003) with a longer disease duration (Fig. 1D, P=0.05) compared to other samples. Group 1 samples tended to have worse pulmonary hypertension (Fig. 1E, P=0.004) and were less likely to have pulmonary fibrosis (Fig. 1F, P=0.05) than the remainder of samples. Lastly, a regulator interaction network was created to provide novel insights for SSc and visualize the connectivity between different components of the regulator network.
Conclusion:
We have identified a subtype of samples classified as the inflammatory gene expression subset that show increased expression of innate immune pathways NFAT, NFKB and STAT3. Patients with high expression of two of these pathways, which we have termed Ôdouble highÕ, had evidence of increased disease severity. This further highlights the role of innate immune transcriptional activators in SSc pathogenesis. The framework can be easily applied to any samples with molecular gene expression data to develop transcriptional activity networks.
To cite this abstract in AMA style:
Wang Y, Franks J, Whitfield ML. Identification of Transcriptional Regulatory Networks in Systemic Sclerosis [abstract]. Arthritis Rheumatol. 2018; 70 (suppl 9). https://acrabstracts.org/abstract/identification-of-transcriptional-regulatory-networks-in-systemic-sclerosis/. Accessed .« Back to 2018 ACR/ARHP Annual Meeting
ACR Meeting Abstracts - https://acrabstracts.org/abstract/identification-of-transcriptional-regulatory-networks-in-systemic-sclerosis/