Session Information
Session Type: Poster Session A
Session Time: 10:30AM-12:30PM
Background/Purpose: Hypersensitivity pneumonitis (HP) is an interstitial lung disease induced by inhaled antigens that trigger an interstitial and bronchoalveolar lymphocytic inflammatory response. This immune response is characterized by the proliferation of cytotoxic CD8+ lymphocytes and the production of specific antibodies (precipitins) by plasma cells, stimulated by CD4+ Th1 lymphocytes. It has been observed that some patients with HP develop autoimmune phenomena.
The scope of this study is to analyze the characteristics, progression, and response to treatment of a cohort of adult patients with HP with autoimmune features (HPAF).
Methods: Retrospective study of patients with HPAF diagnosed in a Functional Unit of Interstitial Lung service of a tertiary-level university hospital. The diagnosis of HP was established based on the ATS/JRS/ALAT consensus criteria of 2020.
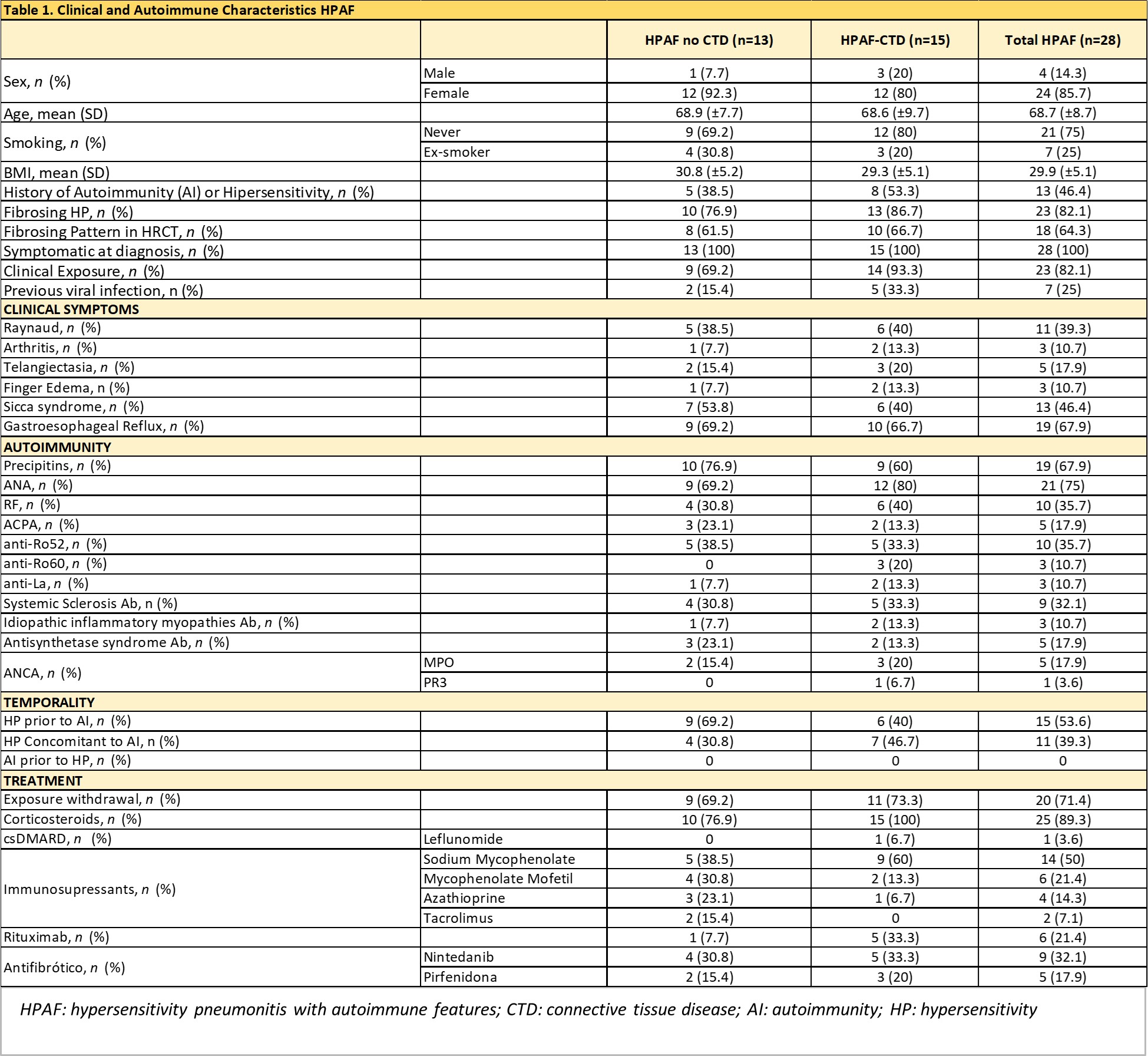
Results: To date, 28 patients have been identified, with an average age at diagnosis of 68.7±8 years. The main characteristics of these patients are detailed in Table 1.
Twenty-three (82%) had a fibrosing ILD, being NSIP the most common radiological pattern (64.2%).
Fifteen patients (53.5%) were diagnosed with connective tissue diseases (CTD), including systemic sclerosis (7), Sjögren’s syndrome (4), ANCA vasculitis (3), and rheumatoid arthritis (1). The remaining 13 had clinical and/or immunological data suggestive of autoimmunity, but did not meet criteria for CTD.
In 53.6% of the patients, the diagnosis of HP preceded the detection of autoimmunity by an average of 26.8 months; in the remaining 46.4%, the autoimmune disorder was concurrent with the time of diagnosis.
The main clinical manifestations in patients with HPAF were sicca syndrome (53.8%) and Raynaud’s phenomenon (39.3%). The most frequent antibodies were ANA.
Treatment with glucocorticoids was initiated in 89.3% of patients, immunosuppressive agents in 75%, rituximab in 21.4%, and antifibrotic treatment in 39.3%.
After a median follow-up of 50 months (IQR 25th–75th: 20.2 – 101.7), clinical worsening was observed in 67.9% of patients, radiological progression in 64.3%, and deterioration in respiratory functional tests in 57.1%. Nine ended up developing chronic respiratory failure requiring oxygen therapy, 6 were candidates for lung transplantation, and 3 died (after a median follow-up of 104.4 months).
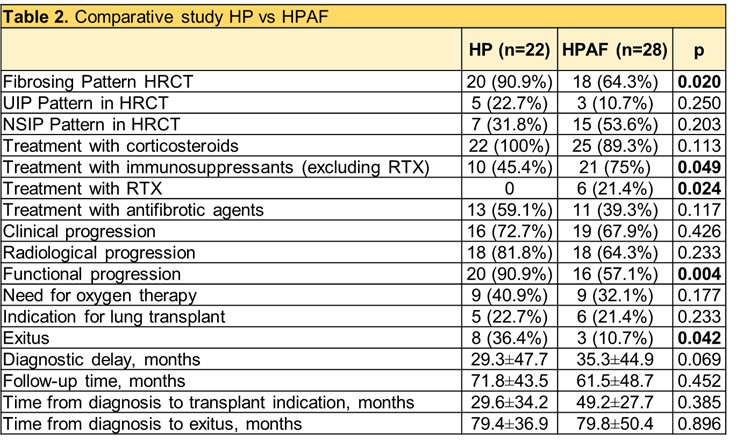
In the comparative study (Table 2), patients with HPAF had less functional deterioration (57.1% vs. 90.9%, p=0.004) and lower mortality (10.7% vs. 36.4%, p=0.042) compared to controls with HP without autoimmunity.
Conclusion: In clinical practice, it is common to identify a subgroup of patients with HP and autoimmune characteristics. HP and various CTD share a dysregulation of T cells, which could explain the higher incidence of autoimmune disease in patients with HP. It remains to be determined whether HPAF is a distinct clinical phenotype of HP. In our experience, immunosuppressive treatment in these patients seems to reduce functional deterioration and decrease mortality, similarly to IPAF. The lower proportion of fibrosing patterns in HPAF could explain its effectiveness.
To cite this abstract in AMA style:
Maymó P, Narvaez-García J, Bermudo G, Vicens-Zygmunt V, Bolivar S, del Río B, Palacios J, Aguilar Coll M, Roig Kim M, De Daniel L, Vidal P, Nolla J, Molina M. Hypersensitivity Pneumonitis with Autoimmune Features: A New Entity? [abstract]. Arthritis Rheumatol. 2024; 76 (suppl 9). https://acrabstracts.org/abstract/hypersensitivity-pneumonitis-with-autoimmune-features-a-new-entity/. Accessed .« Back to ACR Convergence 2024
ACR Meeting Abstracts - https://acrabstracts.org/abstract/hypersensitivity-pneumonitis-with-autoimmune-features-a-new-entity/