Session Information
Date: Monday, October 27, 2025
Session Type: Poster Session B
Session Time: 10:30AM-12:30PM
Background/Purpose: Lymphocytic interstitial pneumonia (LIP) is a rare interstitial lung disease, most commonly associated with Sjögren’s syndrome, but it may also occur in the context of other autoimmune diseases. Despite its low prevalence, LIP can significantly impact patients’ quality of life, particularly in cases progressing to severe pulmonary restriction. Accurate characterization of LIP is essential, as early diagnosis can guide timely interventions, including consideration for lung transplantation in advanced cases.
Methods: We conducted a cross-sectional study of patients with lymphocytic interstitial pneumonia (LIP) diagnosed by multidisciplinary consensus between rheumatology and pulmonology. Clinical, serological, and functional variables were systematically collected. Pulmonary function tests included forced vital capacity (FVC); a restrictive pattern was defined as FVC < 80% of predicted. Chest CT scans were evaluated for the number and size of pulmonary cysts. Cystic disease was considered diffuse when more than 10 cysts were identified. The largest cyst in each patient was measured along its longest axis and reported in millimeters.
Results: A total of 46 patients with LIP were included; 58.7% had associated Sjögren’s syndrome. Other autoimmune diagnoses included rheumatoid arthritis (10.9%), rheumatoid arthritis with Sjögren’s overlap (6.5%), systemic sclerosis (8.7%), lupus (2.2%), antisynthetase syndrome (2.2%), and interstitial pneumonia with autoimmune features (IPAF) (2.2%). Four patients (8.7%) had no identifiable autoimmune disease.There were no significant differences in age, sex, smoking history, or pulmonary function between groups. Sjögren’s patients had a higher prevalence of ANA positivity (100% vs 78.6%, p=0.01), anti-Ro (71.4% vs 25%, p< 0.01), and anti-La antibodies (65% vs 20%, p< 0.05). High-titer anti-Ro was significantly more frequent in the Sjögren’s group (61.9% vs 18.8%, p< 0.05). Serum immunoglobulin levels, complement levels, and the proportion of patients with IgG >2000 mg/dL did not differ significantly. Although diffuse cystic disease (defined as >10 cysts) was common in both groups (88.9% vs 73.7%, p=0.18), the largest cyst size was significantly greater in patients with Sjögren’s (33.8 ± 26.0 mm vs 19.8 ± 10.6 mm, p=0.03).
Conclusion: In this cohort of patients with LIP, those with Sjögren’s syndrome exhibited a more pronounced autoimmune profile and significantly larger cysts, despite similar lung function. These findings support the hypothesis that LIP in Sjögren’s may represent a distinct immunopathologic phenotype, potentially driven by exaggerated bronchus-associated lymphoid tissue (BALT) hyperplasia. Early recognition of this phenotype may inform prognosis and guide decisions regarding close monitoring or early referral for lung transplantation.
 Comparison of Clinical, Serological, and Radiological Characteristics in Patients With Lymphocytic Interstitial Pneumonia According to Sjögren’s Status
Comparison of Clinical, Serological, and Radiological Characteristics in Patients With Lymphocytic Interstitial Pneumonia According to Sjögren’s Status
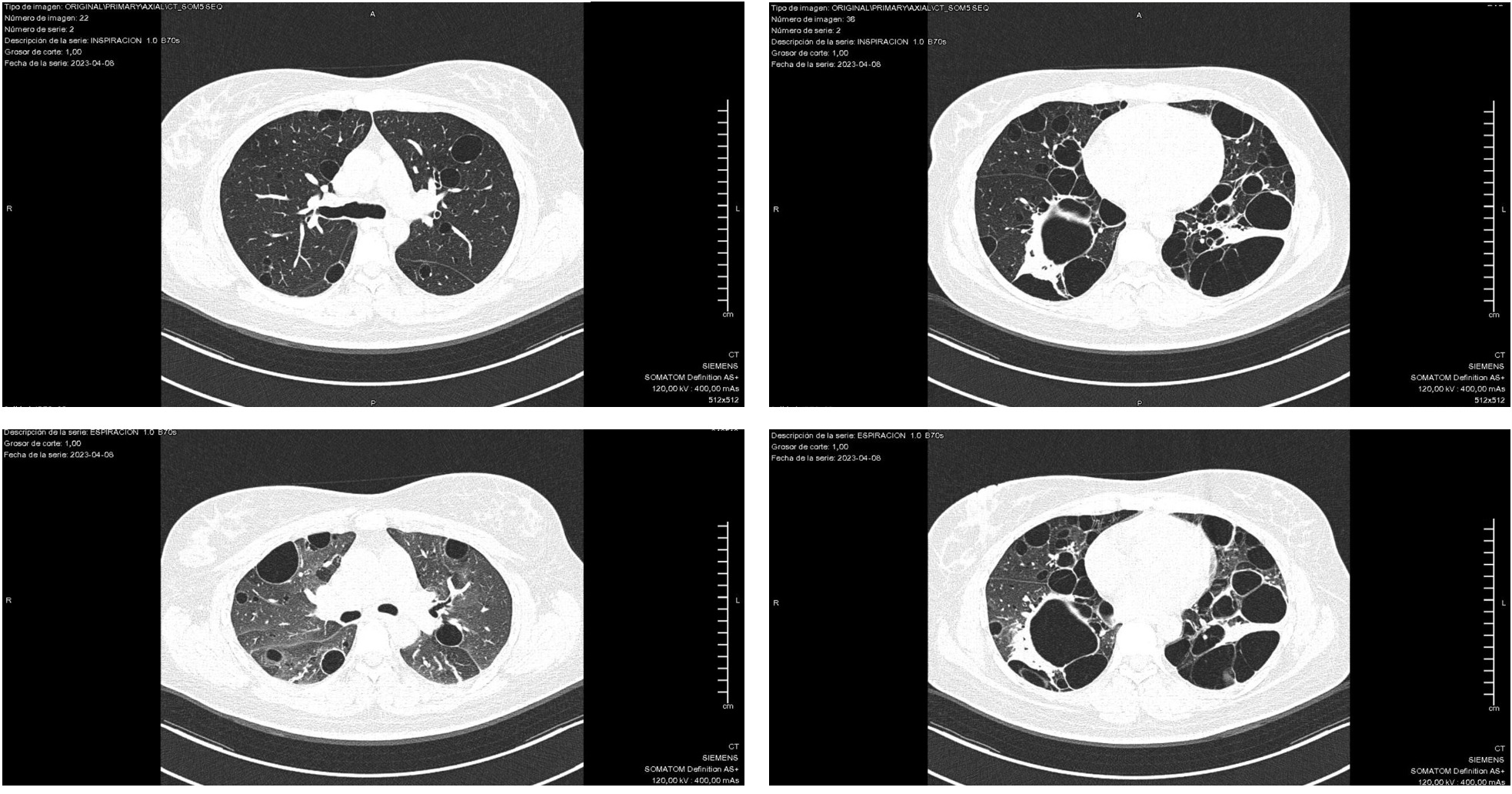
.jpg) Chest CT of a Patient With Sjögren’s Syndrome and Lymphocytic Interstitial Pneumonia Showing Cysts >10 mm in Diameter; A–B: Inspiratory Phase, C–D: Expiratory Phase
Chest CT of a Patient With Sjögren’s Syndrome and Lymphocytic Interstitial Pneumonia Showing Cysts >10 mm in Diameter; A–B: Inspiratory Phase, C–D: Expiratory Phase
To cite this abstract in AMA style:
CHACON K, MARQUEZ-SANCHEZ A, Rojas-Serrano J, MEJÍA M. Cystic Lung Disease and Autoantibodies in Lymphocytic Interstitial Pneumonia: Exploring the Impact of Sjögren’s Syndrome [abstract]. Arthritis Rheumatol. 2025; 77 (suppl 9). https://acrabstracts.org/abstract/cystic-lung-disease-and-autoantibodies-in-lymphocytic-interstitial-pneumonia-exploring-the-impact-of-sjogrens-syndrome/. Accessed .« Back to ACR Convergence 2025
ACR Meeting Abstracts - https://acrabstracts.org/abstract/cystic-lung-disease-and-autoantibodies-in-lymphocytic-interstitial-pneumonia-exploring-the-impact-of-sjogrens-syndrome/