Session Information
Date: Monday, November 11, 2019
Title: Systemic Sclerosis & Related Disorders – Basic Science Poster
Session Type: Poster Session (Monday)
Session Time: 9:00AM-11:00AM
Background/Purpose: Clinical trials with systemic sclerosis (SSc) patients have yet to lead to an FDA approved treatment. We have adopted a gene fold-change analysis called “IDEA” to compare cell lines from the CMAP 2.0 database with biopsies of SSc patients. Using IDEA, we can identify perturbagens that induce a genetic signature in cell lines to be more comparable with healthy controls than SSc patients. We have also created correlation networks with patient cohort gene expression data. Head nodes within these networks may be targetable via perturbagens, disrupting genes highly correlated with the SSc disease phenotype. Here, we use this method to identify several potential perturbagens that may modulate the SSc phenotype.
Methods: Raw .cel files were downloaded from CMAP 2.0, processed with RMA, quantile normalized and fit to a multichip linear model. Probes are collapsed at average intensity while gene fold-change is the ratio of treatment to control intensities followed by a log2-transformation. The data is then processed for use with the BASE algorithm. A t-test (Bonferroni corrected) is calculated to determine which perturbagens have a gene signature most similar to healthy controls when compared to either the Inflammatory or Fibroproliferative SSc patient subsets. Patient cohort data is run through WGCNA using a signed network. Modules that correlate highly with patient subsets are chosen to create their respective correlation networks.
Results: Using two independent microarray datasets, we found four overlapping perturbagens that affect the inflammatory subset and fourteen that affect the fibroproliferative subset. Using drug set enrichment analysis (DSEA), we were able to classify which drugs are more highly enriched in cell signaling pathways and immune system pathways. PI3K-inhibitors and an immunosuppressive highly regulate these pathways. WGCNA was used to create correlation networks based on genes most associated with patient gene expression subtypes. The top hub nodes for the inflammatory network were MS4A6A, CD93 and HLA-DMA, while the fibroproliferative network had IKBKG (fig. 1). These hubs and their associated genes may be key gene expression hubs to target with for intervention.
Conclusion: We have identified a wide range of perturbagens ranging in mechanism of action, from anti-inflammatories to anti-psychotics and anti-biotics as potential therapeutic perturbagens of interest to further test. These perturbagens have been shown to genetically modulate the molecular pathways dysregulated in SSc, opening a different avenue towards treating the disease.
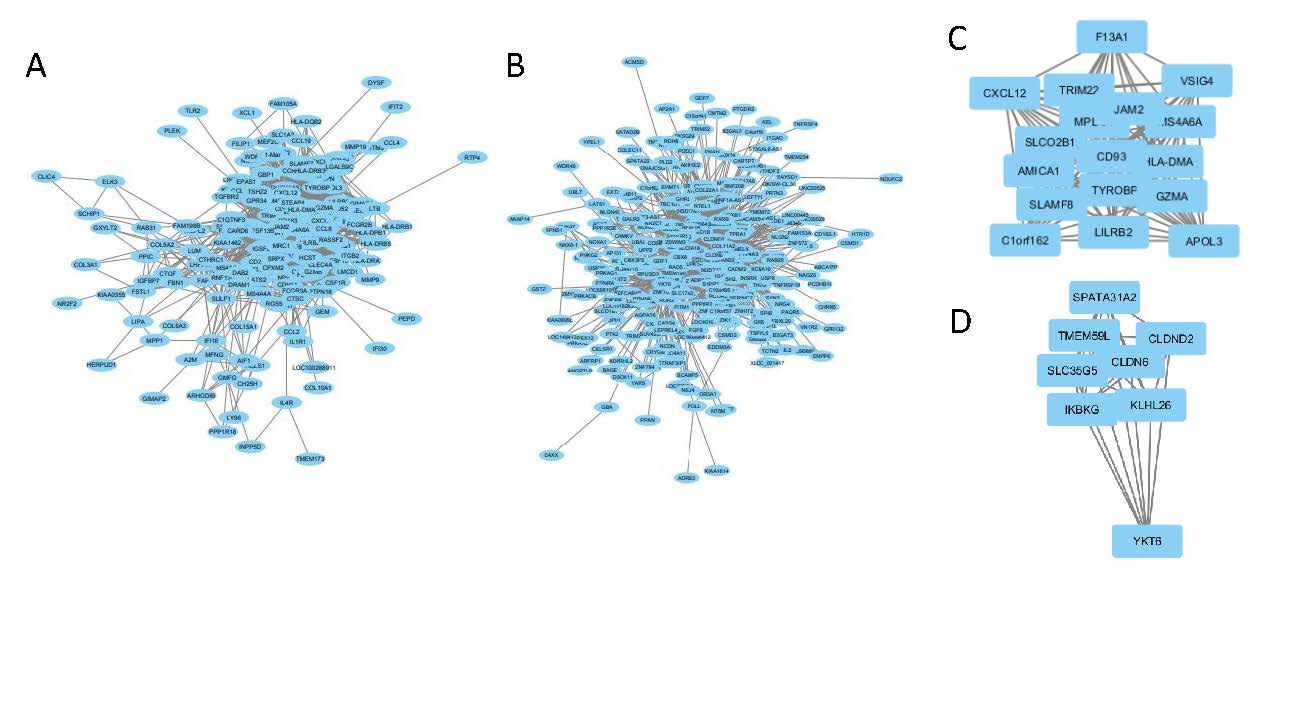
Figure1
To cite this abstract in AMA style:
Popovich D, Whitfield M, Wang Y, Cai G, Huang M. Computational Methods for Drug Repositioning of Systemic Sclerosis Using Gene Fold-Change and Network Analyses [abstract]. Arthritis Rheumatol. 2019; 71 (suppl 10). https://acrabstracts.org/abstract/computational-methods-for-drug-repositioning-of-systemic-sclerosis-using-gene-fold-change-and-network-analyses/. Accessed .« Back to 2019 ACR/ARP Annual Meeting
ACR Meeting Abstracts - https://acrabstracts.org/abstract/computational-methods-for-drug-repositioning-of-systemic-sclerosis-using-gene-fold-change-and-network-analyses/