Session Information
Session Type: Poster Session C
Session Time: 9:00AM-11:00AM
Background/Purpose: Interstitial lung disease (ILD) is a common extra-muscular manifestation of Idiopathic Inflammatory Myositis (IIM) and increases risk of mortality. Prior studies and registries have focused on either smaller cohorts or predominantly Caucasian/European populations. Our aim was to better characterize the clinical disease manifestations of ILD in racially diverse population of IIM patients in the Bronx, NY, particularly to describe the associated phenotypes and antibody profiles, clinical skin manifestations, and autoimmune co-morbidities.
Methods: Montefiore Medical Center patients that met 2017 EULAR/ACR classification criteria for IIM were included. The presence of ILD, as well as the radiographic pattern, severity, and progression were determined using available pulmonary function testing and CT of the chest. Statistical analyses included Chi-Square test and Fisher’s test.
Results: There were 152 myositis patients in this cohort, of which 62 patients had evidence of ILD (41%). Among the 62 patients with ILD, 71% were women and 29% were men. Patients with ILD were 44% Latin ethnicity and consisted of 56% Black, 24% White, 3% American Indian, 3% Asian (Table 1). Patients with ILD were radiographically classified as follows: 18 (29%) with ground glass opacities without specific pattern, 6 (10%) with usual interstitial pneumonia (UIP), 21 (34%) with nonspecific interstitial pneumonia (NSIP), and 17 (27%) with other patterns of ILD (Table 2).
Among patients with ILD, 22 (35%) had PM, 38 (61%) had DM with 7 of those patients having CADM, 2 (3%) had IBM, and 1 (2%) patient had necrotizing (statin-induced) myopathy. Of the PM/DM patients, 14 (23%) qualified as having anti-synthetase syndrome and 13 (21%) had MCTD, UCTD, or an overlap syndrome, both of which had higher ILD prevalences (p=0.0002 and p=0.0001). There was no statistical associated between IBM and ILD (p value = 0.01) but there were 2 cases of IBM with ILD. Patients with ILD had higher prevalence of Jo-1 and SSA antibodies (p=0.002, p=0.003). Dermatomyositis patients with Mi-2 antibody had no prevalence of ILD in this cohort (p=0.03) (Table 3). Majority of these findings are similar to larger, predominant Caucasian studies of IIM.
While IIM patients with ILD showed varying skin manifestations, there were no particular skin manifestations that were significantly more prevalent in patients with ILD in this racially diverse cohort. Patients with ILD also had a higher prevalence of one or more autoimmune comorbidities (p=0.01). Conversely, patients without ILD had higher prevalence of no autoimmune co-morbidities (p=0.04).
Conclusion: ILD in this racially diverse population with IIM is a significant manifestation of disease that presents heterogeneously. This study serves to confirm that certain myositis phenotypes, associated antibody profiles, and autoimmune comorbidities are associated with ILD, even in a racially diverse urban population. Limitations include a small sample size of antibody profiles for study. Future direction will include further stratifying patients with ILD in this diverse population by radiographic patterns to assess if response to medications is similar to more homogenous cohorts.
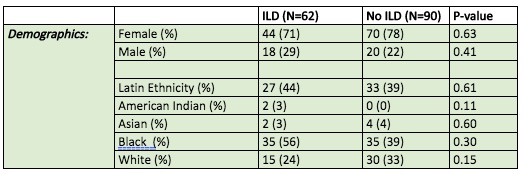 Table 1: Patient Demographics
Table 1: Patient Demographics
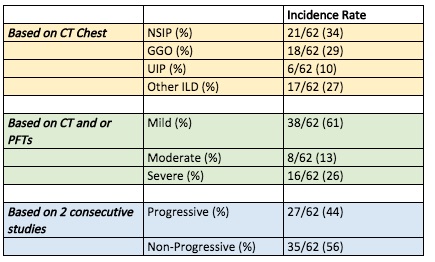 Table 2: Incidence of ILD subtypes in IIM patients
Table 2: Incidence of ILD subtypes in IIM patients
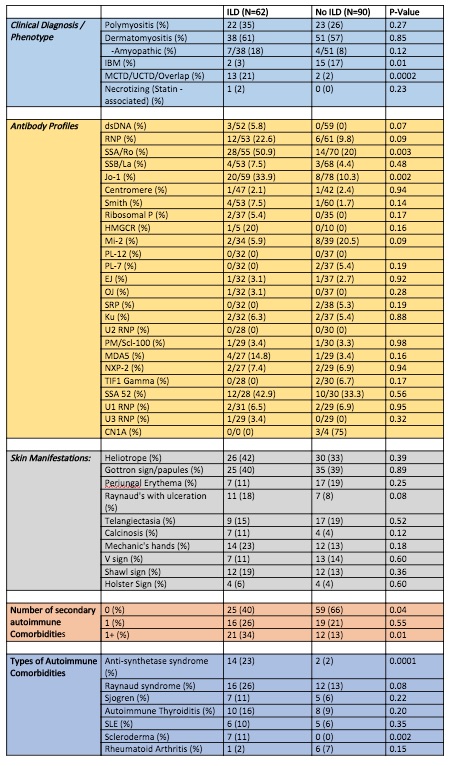 Table 3: Antibody profiles, clinical manifestations, and autoimmune co-morbidities in IIM Patients with ILD
Table 3: Antibody profiles, clinical manifestations, and autoimmune co-morbidities in IIM Patients with ILD
To cite this abstract in AMA style:
Law J, Valle A, Mullins K, Mahmood S. Assessing Interstitial Lung Disease in a Racially Diverse Population with Idiopathic Inflammatory Myositis [abstract]. Arthritis Rheumatol. 2020; 72 (suppl 10). https://acrabstracts.org/abstract/assessing-interstitial-lung-disease-in-a-racially-diverse-population-with-idiopathic-inflammatory-myositis/. Accessed .« Back to ACR Convergence 2020
ACR Meeting Abstracts - https://acrabstracts.org/abstract/assessing-interstitial-lung-disease-in-a-racially-diverse-population-with-idiopathic-inflammatory-myositis/