Session Information
Session Type: Poster Session A
Session Time: 9:00AM-11:00AM
Background/Purpose: Sjögren’s Disease (SjD) has high glandular IFNg levels, associated with disease activity and lymphoma risk. We showed that IFNg-stimulated minor salivary gland (SG)-mesenchymal stromal cells (MSCs) produced CXCL-9, -10, and -11 through JAK2→STAT1 phosphorylation. Chemokine production and STAT1 phosphorylation by SG-MSCs were inhibited by the JAK1/2 inhibitor, ruxolitinib, a potential promising therapy for SjD. Using culture adapted SG MSC as an in vitro rosetta stone of SG-resident parenchymal cells, we investigated the IFNg →JAK/STAT biochemical response of SG-MSCs to FDA approved JAK inhibitors.
Methods: We treated human SG-MSCs with IFNg and JAK1/2 inhibitors. The JAK inhibitors included ruxolitinib and baricitinib, which bind the JAK conformation poised to phosphorylate its STAT substrates (kinase active conformation). We also studied the novel JAK inhibitor, CHZ868, which binds the kinase inactive JAK conformation. We performed western blotting of cell lysates, probing for JAK1/2 or STAT1/3. We performed RNA sequencing and confirmed results with qPCR and ELISA. ANOVA or paired t-tests were used to calculate p-values across multiple variables.
Results: We determined that ruxolitinib treatment increased phospho JAK1 (pJAK1) and phospho JAK2 (pJAK2) of IFNg-stimulated SG-MSCs in a dose-dependent manner (Fig 1a-b). pSTAT1 and pSTAT3 were suppressed by ruxolitinib upon treatment (Fig 1c-d). Discontinuation of ruxolitinib resulted in decreased pJAK1/2 (Fig1e). We found a marked and rapid increase of pSTAT1 upon discontinuation of ruxolitinib and baricitinib, but not CHZ868 (Fig 1f). Next, we wanted to determine the downstream effects of this pSTAT cascade. We performed RNA-seq of three conditions: 1) IFNg throughout; 2) IFNg with ruxolitinib, discontinue ruxolitinib, and collect conditioned media 3 hours later; 3) IFNg with CHZ868, discontinue CHZ868, and collect conditioned media 3 hours later. We found that ruxolitniib withdrawal was associated with increased PLAT and PLAU expression when compared to IFNg treatment or CHZ868 discontinuation (Fig 2a-b). We confirmed that PLAU (plasminogen activator, urokinase [uPA]) increased in response to ruxolitinib discontinuation but not in control or CHZ868 discontinuation by qPCR (Fig 3a-b) and ELISA (Fig 3c).
Conclusion: Our findings suggest that ruxolitinib and baricitinib bind the active phosphorylated form of JAK and lead to a paradoxical cellular accumulation of functionally defective pJAK. Upon inhibitor withdrawal, the primed pJAKs are de-repressed and initiate a pSTAT signaling cascade, ultimately resulting in high levels of uPA. In contrast, CHZ868, which binds the inactive JAK kinase conformation, does not lead to pJAK accumulation, a pSTAT cascade, or uPA production. CHZ868 does not cause this phenomenon. High uPA, associated with human atherosclerotic plaques, might cause proteolysis and plaque rupture. In conclusion, JAK1/2 inhibitors that bind the active pJAK configuration, but not those that bind the inactive JAK configuration, increase STAT signaling on withdrawal. This signaling results in high uPA and might promote plaque rupture (Fig 3d).
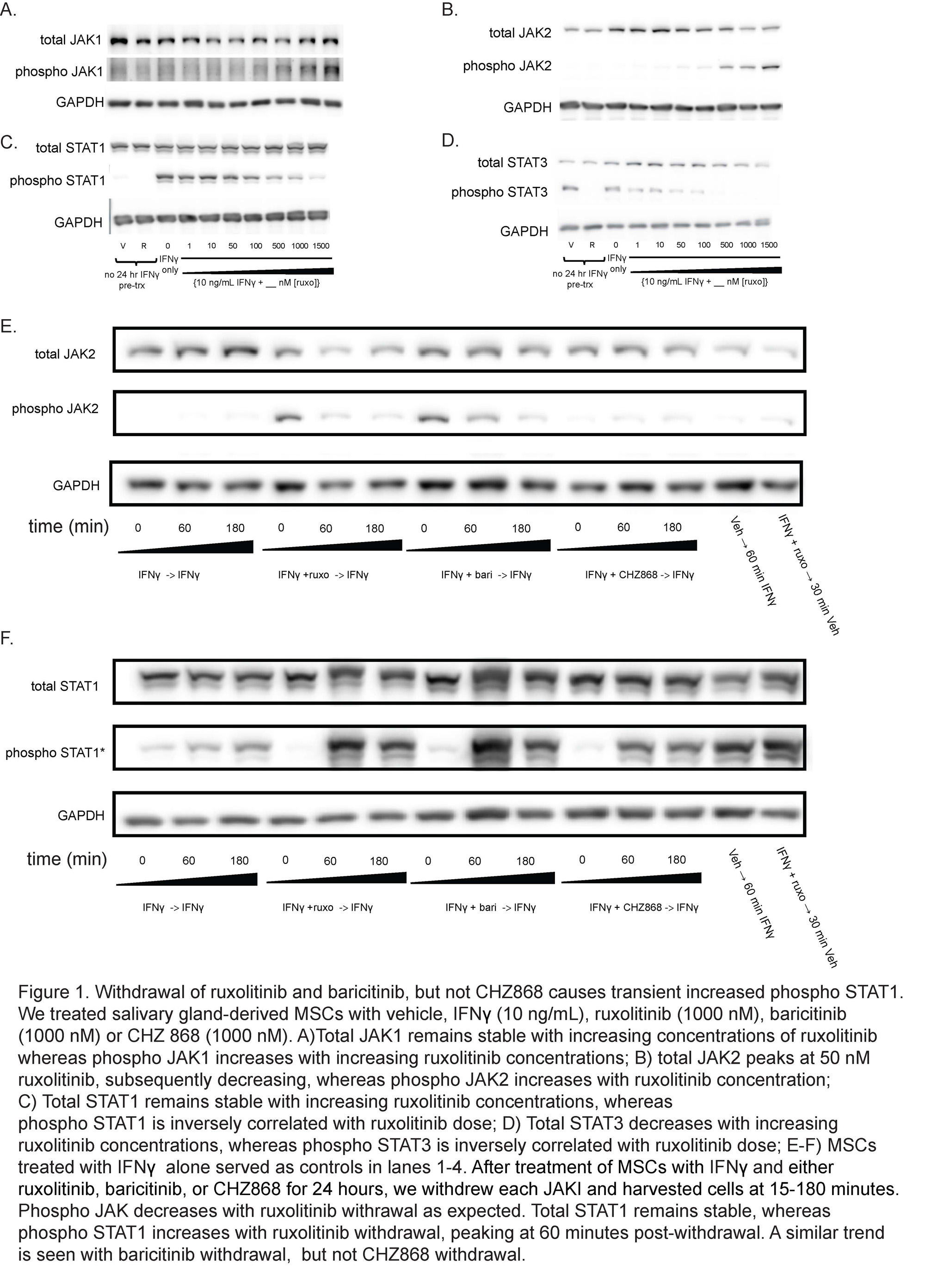
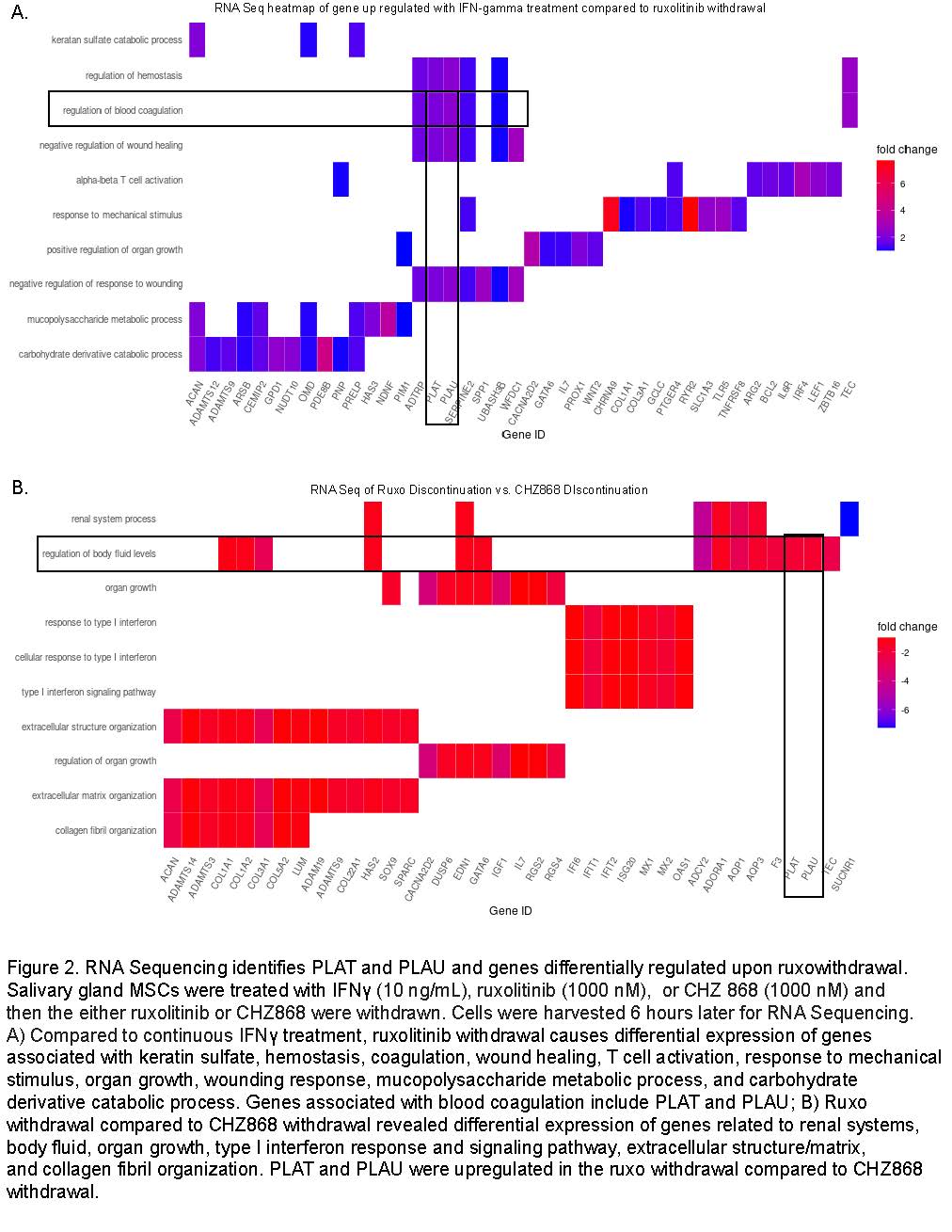
Salivary gland MSCs were treated with IFNγ (10 ng/mL), ruxolitinib (1000 nM), or CHZ 868 (1000 nM) and
then the either ruxolitinib or CHZ868 were withdrawn. Cells were harvested 6 hours later for RNA Sequencing.
A) Compared to continuous IFNγ treatment, ruxolitinib withdrawal causes differential expression of genes associated with keratin sulfate, hemostasis, coagulation, wound healing, T cell activation, response to mechanical stimulus, organ growth, wounding response, mucopolysaccharide metabolic process, and carbohydrate derivative catabolic process. Genes associated with blood coagulation include PLAT and PLAU; B) Ruxo withdrawal compared to CHZ868 withdrawal revealed differential expression of genes related to renal systems, body fluid, organ growth, type I interferon response and signaling pathway, extracellular structure/matrix, and collagen fibril organization. PLAT and PLAU were upregulated in the ruxo withdrawal compared to CHZ868 withdrawal.

To cite this abstract in AMA style:
McCoy S, Gurevic I, Galipeau j. A Potential Mechanism for Major Adverse Cardiac Events Associated with JAK Inhibitors: JAK Inhibitor Withdrawal Causes Urokinase Release by Primed STAT Signaling [abstract]. Arthritis Rheumatol. 2023; 75 (suppl 9). https://acrabstracts.org/abstract/a-potential-mechanism-for-major-adverse-cardiac-events-associated-with-jak-inhibitors-jak-inhibitor-withdrawal-causes-urokinase-release-by-primed-stat-signaling/. Accessed .« Back to ACR Convergence 2023
ACR Meeting Abstracts - https://acrabstracts.org/abstract/a-potential-mechanism-for-major-adverse-cardiac-events-associated-with-jak-inhibitors-jak-inhibitor-withdrawal-causes-urokinase-release-by-primed-stat-signaling/