Session Information
Session Type: Poster Session A
Session Time: 9:00AM-11:00AM
Background/Purpose: Gene expression analysis coupled with machine learning (ML) holds the promise of identifying subsets of patients with heterogeneous diseases, such as systemic lupus erythematosus (SLE), a multi-organ autoimmune disease with known diversity in both clinical presentation and gene expression profiles. However, phenotype prediction based on gene expression has often relied on the performance of individual genes without a systems biology context. To address this, we created an interpretable ML approach to predict clinical phenotype of SLE patients in a non-invasive manner using blood transcriptomic profiles.
Methods: We developed a sequential grouped feature importance algorithm to assess the performance of gene sets, including those identifying immune and metabolic pathways and cell types known to be abnormal in SLE, in predicting the presence of lupus as well as disease activity and organ involvement. We normalized, merged, and batch-corrected publicly available datasets and created six ML models to predict SLE from healthy controls (CTL), inactive SLE from CTL, active from inactive SLE, lupus nephritis (LN) from CTL, LN from non-renal lupus, and rheumatoid arthritis (RA) from SLE.
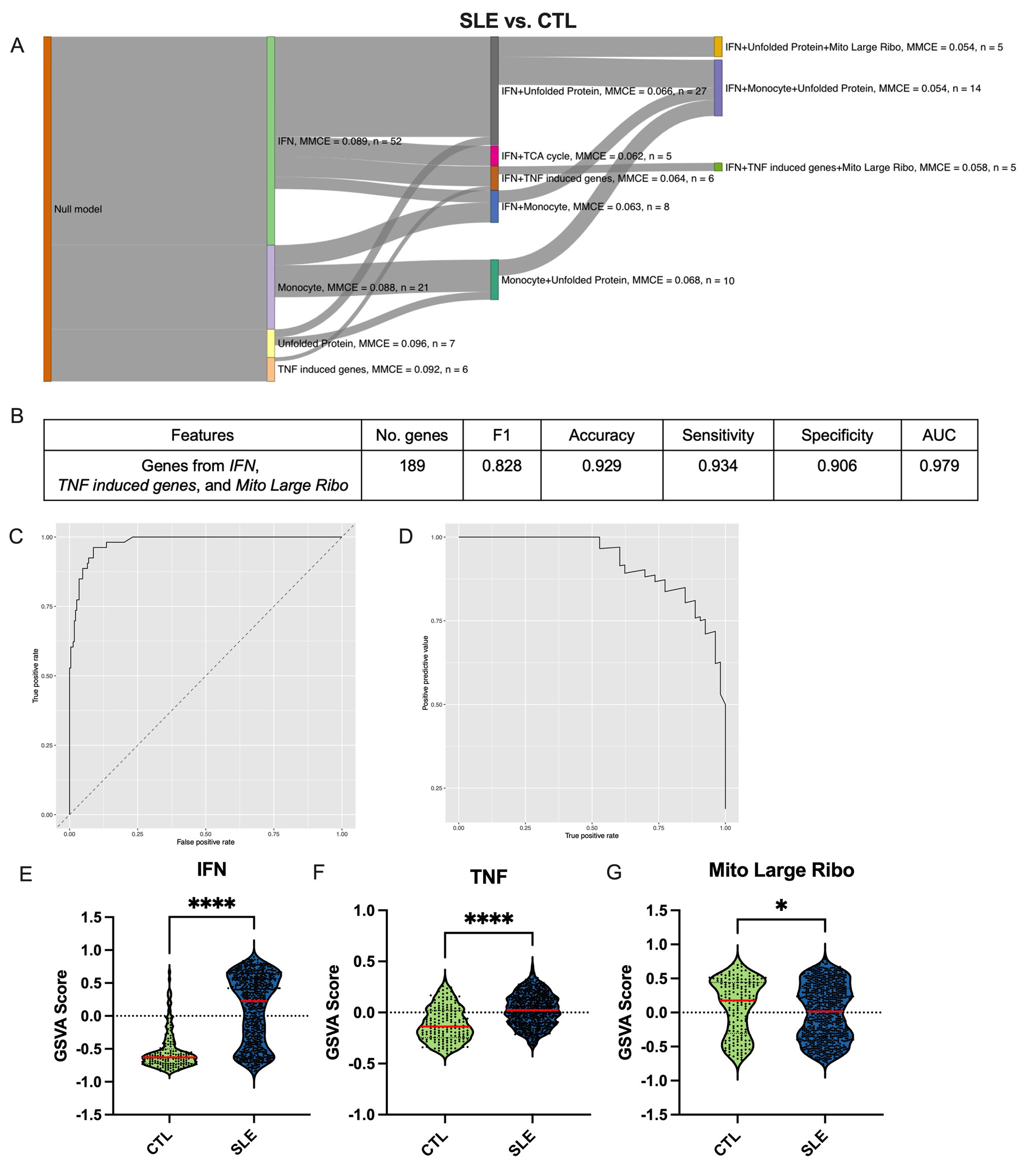
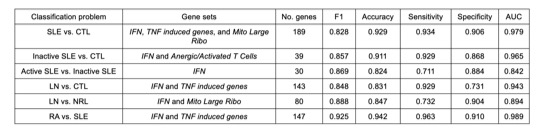
Results: The SGFI algorithm first selects the best gene set to predict phenotype for the model, and then sequentially adds in additional gene sets if they improve the performance of the model (Figure 1A). SGFI provides a way to reduce the high dimensionality of transcriptomic datasets meaningfully, as it incorporates prior knowledge of biology into the data while also selecting pathways in an unbiased manner, leading to biologically informative conclusions. After feature selection, the best gene set combination was found via 10-fold cross validation on the train set and then evaluated on the test set (Figure 1B-D). We then performed gene set variation analysis to examine how these pathways differ across clinical phenotypes (Figure 1E). Gene sets related to interferon, tumor necrosis factor, the mitoribosome, and anergic/activated T cell were the best predictors of phenotype in all classifications (Table 1). The ML models created with those genes as features had excellent performance with AUCs ranging from 0.842 to 0.989 and accuracies of 0.824 to 0.942 (Table 1).
Conclusion: A novel feature selection approach combined with interpretable ML performs extremely well in predicting SLE phenotypes and in separating patients with lupus from both normal and those with RA. Moreover, since interpretable ML can be used to suggest potential causal relationships, these results point to associations between the molecular pathways identified in each model and manifestations of SLE pathogenesis. This innovative ML approach can help improve recognition of SLE phenotypic subsets and additionally be applied to other diseases and tissues.
To cite this abstract in AMA style:
Leventhal E, Daamen A, Grammer A, Lipsky P. A Novel Transcriptome-Based Machine Learning Pipeline Predicts Phenotypes of Lupus Patients [abstract]. Arthritis Rheumatol. 2023; 75 (suppl 9). https://acrabstracts.org/abstract/a-novel-transcriptome-based-machine-learning-pipeline-predicts-phenotypes-of-lupus-patients/. Accessed .« Back to ACR Convergence 2023
ACR Meeting Abstracts - https://acrabstracts.org/abstract/a-novel-transcriptome-based-machine-learning-pipeline-predicts-phenotypes-of-lupus-patients/