Session Information
Session Type: ACR Concurrent Abstract Session
Session Time: 4:30PM-6:00PM
Background/Purpose: Detecting distinct cellular subsets in disease tissues is key to understanding the pathogenesis of immune diseases, for example in synovial tissues in rheumatoid arthritis (RA). Yet this task is complicated by the highly heterogeneous nature of the immune system. Recently, advances in single-cell technologies have enabled us to increase the power to resolve this heterogeneity to find disease relevant and cell type subsets. Integration of single-cell data with bulk data will help identify differentially abundant cell subsets.
Methods: We develop a novel statistical method for integrative analysis of any single-cell assay (flow cytometry, single-cell RNA-seq, or mass cytometry) with bulk RNA-seq (Figure 1). Our method has three steps. (1) We find the principal components (PCs) in the bulk RNA-seq data. (2) We model cell type abundances as a linear combination of bulk PCs. With single-cell data, we model a single-cell measurement with each PC to identify important signatures of bulk RNA-seq. The identified subsets are reflected as distinct clusters of cells in single-cell data and explain the variance in the bulk data. In mass cytometry data, we use a logistic model that includes the PC as a variable to predict the cluster identity of a cell. (3) To assess significance, we use permutations to test statistical significance of model coefficients.
Results: Using this integrative method, we are able to identify cellular subsets and statistically significant marker genes (permutation P < 1e-5) in fibroblasts, B cells, and T cells in AMP Phase 1 RA data (Bulk RNA-seq: 40 RA and 14 OA; Single-cell RNA-seq: 22 RA and 3 OA; Mass cytometry: 15 RA and 11 OA). Synovial fibroblasts separate into lining fibroblasts marked by CD55 and PRG4, and sublining fibroblasts marked by THY1 and CD74. B cells were separated into two distinct subsets: activated B cells that express HLA-DRA, CD37, and CD83 and plasma cells that express XBP1 and FKBP11. T cells were also separated into CD4+ helper that express CD4, IL7R, and SELL and CD8+ cytotoxic that express CD8A, CCL5, and HLA-DQA1. We tested subsets of these populations for disease association. We found that fibroblast subset marked by THY1 were significantly associated with RA (logistic regression P < 2e-5) when we integrated bulk data with mass cytometry. Derived from the bulk PCs, we observed that a PD1+ subset of CD4 memory T cells which have been recently described as peripheral T helper cells also expanded in RA (P < 0.01).
Conclusion: This computational strategy integrates multiple transcriptomic data types from fresh synovial tissue, giving us a view of the cellular heterogeneity relevant to RA. This method may also have promise in integrating single-cell and bulk data from other sources, for example kidney biopsies from systemic lupus erythematosus (SLE) or tumor biopsies. Acknowledgements: We acknowledge AMP RA/SLE network and AMP funding NIH UH2AR067677-01.
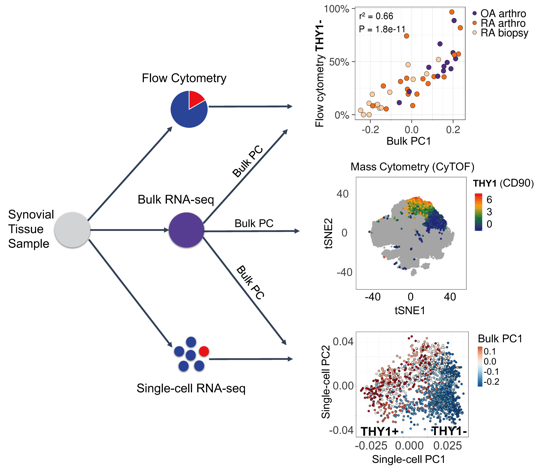
Figure 1. Overview of the data integration method.
To cite this abstract in AMA style:
Zhang F, Slowikowski K, Fonseka C, Wei K, Gutierrez-Arcelus M, Lederer J, Hacohen N, Bykerk VP, Holers M, Gregersen P, McGeachy MJ, Moreland LW, Filer A, Pitzalis C, Lee YC, Anolik JH, Brenner M, Raychaudhuri S. A Novel Statistical Method to Resolve Cellular Heterogeneity in Disease Tissues: Integrating Transcriptomic Data in Accelerating Medicines Partnership (AMP) – RA Network Phase 1 Data [abstract]. Arthritis Rheumatol. 2017; 69 (suppl 10). https://acrabstracts.org/abstract/a-novel-statistical-method-to-resolve-cellular-heterogeneity-in-disease-tissues-integrating-transcriptomic-data-in-accelerating-medicines-partnership-amp-ra-network-phase-1-data/. Accessed .« Back to 2017 ACR/ARHP Annual Meeting
ACR Meeting Abstracts - https://acrabstracts.org/abstract/a-novel-statistical-method-to-resolve-cellular-heterogeneity-in-disease-tissues-integrating-transcriptomic-data-in-accelerating-medicines-partnership-amp-ra-network-phase-1-data/