Session Information
Session Type: Abstract Session
Session Time: 1:00PM-1:15PM
Background/Purpose: COPA syndrome is an autosomal dominant disease caused by missense mutations within the COPA gene. Typical onset begins during childhood with patients developing lung disease such as diffuse alveolar hemorrhage (DAH). Despite its severity, the cause of lung involvement in COPA syndrome is unknown. Here, we find that mutant COPA worsens and prolongs disease in a murine model of DAH and permits aberrant NETosis in humans and mice, offering a route by which lung disease may progress in patients with COPA syndrome.
Methods: Copa+/+ (WT), CopaR233H/+, and CopaR233H/R233H C57BL/6 mice were generated and treated with pristane to induce DAH. Tissues were isolated at 4, 14, or 21 days and analyzed by flow cytometry, ELISA, qPCR, immunofluorescence (IF), and H&E. Circulating whole blood, neutrophils, and plasma were isolated from healthy control (HC) and COPA patients and analyzed by IF, ELISA, and scRNA-seq. Primary lung basal epithelial cells (BECs) isolated from COPA lung explants and immortalized HC BECs were transfected with dsDNA and analyzed via qPCR.
Results: Unlike WT, mutant mice failed to recover from pristane-induced disease as observed by survival, development of DAH, and lung inflammation (Fig 1A-C). Mutant mice underwent dramatic granulopoiesis reflected in dominant neutrophil infiltrates within the BAL, lung, and peritoneum versus WT (Fig 2A). We found elevated neutrophil extracellular traps (NETs) within mutant BAL, and mutant neutrophils underwent increased NETosis compared to WT (Fig 2B-D). Additionally, scRNA-seq of human neutrophils revealed a highly elevated inflammatory signature in COPA versus HC, corroborated by increased NETosis and circulating NETs in these patients (Fig 3A-C). To ask how neutrophils may be recruited into the lung during COPA, we exposed human BECs to dsDNA to simulate alveolar NETs. COPA BECs generated a robust and prolonged inflammatory response compared to HC, specifically IFNB1 and the neutrophil chemoattractant CXCL10, suggesting an amplified capacity to recruit neutrophils (Fig 3D).
Conclusion: In summary, we found that COPAR233H exacerbates pristane-induced lung disease, is associated with increased NETosis, and permits excessive CXCL10 production in epithelial cells. Taken together, our data detail a route by which lung inflammation in COPA syndrome may be promoted in a feed-forward manner via aberrant NETosis and inflamed alveolar epithelium.
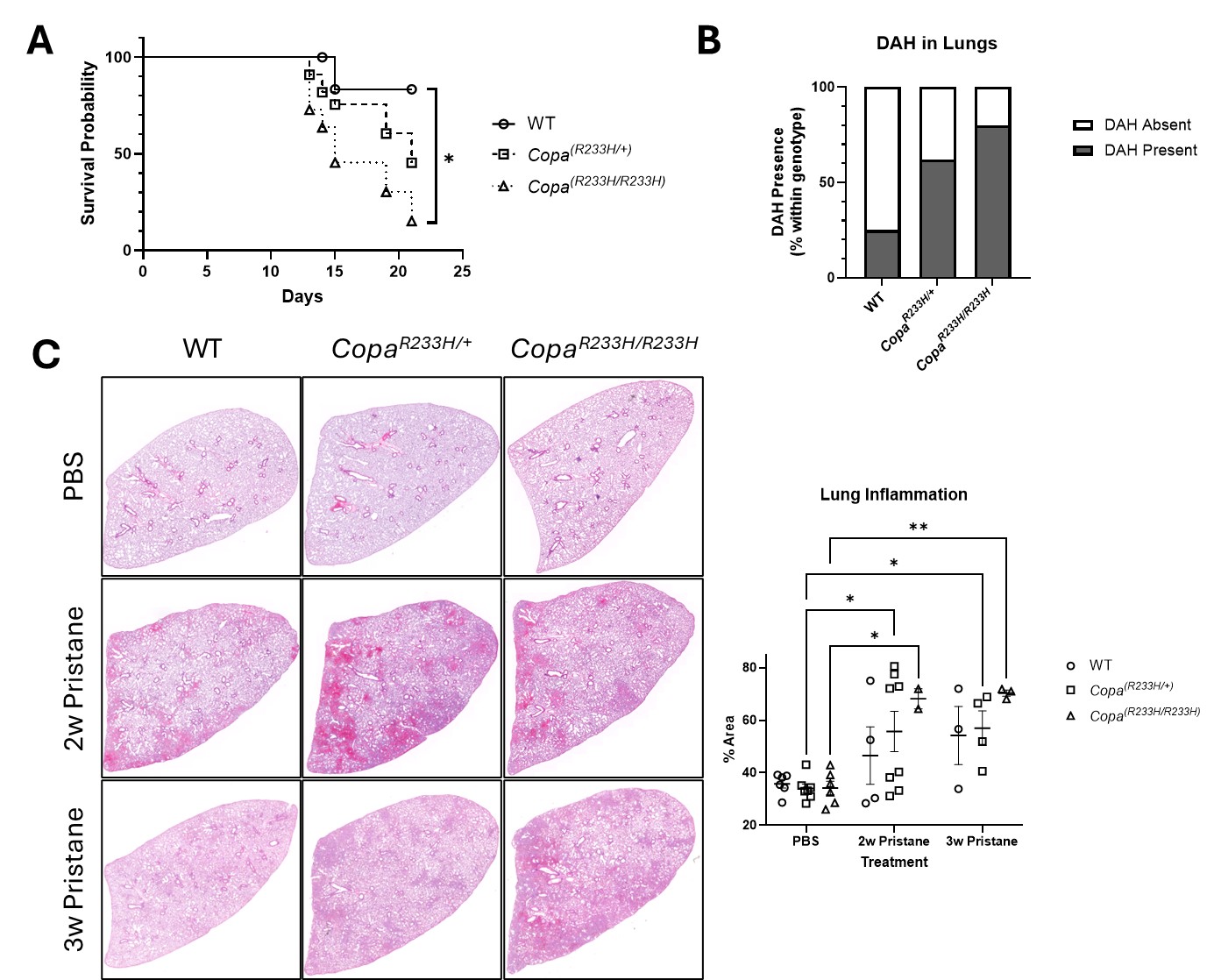 Figure 1: Mice bearing the Copa-R233H allele develop worsened and protracted disease in response to pristane. A) Survival of mice in response to a single intraperitoneal dose of pristane. B) Presence of diffuse alveolar hemorrhage in lungs 2 weeks after pristane injection. C) H&E-stained left lung lobes from PBS- or pristane-treated mice 2 or 3 weeks after i njection (left) as quantified by relative stained area (right). Values are mean +/- SEM. Statistical significance was determined using Log-rank (Mantel-Cox) or Two-Way ANOVA tests. *p < 0.05, **p < 0.01.
Figure 1: Mice bearing the Copa-R233H allele develop worsened and protracted disease in response to pristane. A) Survival of mice in response to a single intraperitoneal dose of pristane. B) Presence of diffuse alveolar hemorrhage in lungs 2 weeks after pristane injection. C) H&E-stained left lung lobes from PBS- or pristane-treated mice 2 or 3 weeks after i njection (left) as quantified by relative stained area (right). Values are mean +/- SEM. Statistical significance was determined using Log-rank (Mantel-Cox) or Two-Way ANOVA tests. *p < 0.05, **p < 0.01.
.jpg) Figure 2: Copa-R233H promotes neutrophil infiltration into inflamed tissue and allows spontaneous and STING-dependent NETosis. A) Neutrophils as a percentage of CD11b+Ly6G+ of CD45+ cells within inflamed tissues 2 weeks after pristane injection. B) Presence of NETs, as measured by citH3::dsDNA complexes, within BAL fluid 2 weeks after pristane injection. C, D) Ability of peritoneal neutrophils to undergo NETosis in response to the STING agonist DMXAA or PMA, as measured by SYTOX+ via flow cytometry (C) and visualized via immunofluorescence (D, arrows). Values are mean +/- SEM. Statistical significance was determined using Two-Way ANOVA. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Figure 2: Copa-R233H promotes neutrophil infiltration into inflamed tissue and allows spontaneous and STING-dependent NETosis. A) Neutrophils as a percentage of CD11b+Ly6G+ of CD45+ cells within inflamed tissues 2 weeks after pristane injection. B) Presence of NETs, as measured by citH3::dsDNA complexes, within BAL fluid 2 weeks after pristane injection. C, D) Ability of peritoneal neutrophils to undergo NETosis in response to the STING agonist DMXAA or PMA, as measured by SYTOX+ via flow cytometry (C) and visualized via immunofluorescence (D, arrows). Values are mean +/- SEM. Statistical significance was determined using Two-Way ANOVA. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
.jpg) Figure 3: COPA-R233H fails to restrain inflammation in patient neutrophils and epithelial cells. A) Neutrophils from patients with COPA syndrome display a dramatic inflammatory signature as determined via scRNA-seq. B, C) Patient neutrophils undergo spontaneous NETosis after 4 hours in culture (B), and patients have elevated circulating NETs as measured by MPO::dsDNA and NE::dsDNA complexes (C). D) COPA patient-derived basal epithelial cells fail to curb IFNB1 and CXCL10 transcript production in response to dsDNA transfection. Values are mean +/- SEM. Statistical significance was determined using Two-Way ANOVA. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Figure 3: COPA-R233H fails to restrain inflammation in patient neutrophils and epithelial cells. A) Neutrophils from patients with COPA syndrome display a dramatic inflammatory signature as determined via scRNA-seq. B, C) Patient neutrophils undergo spontaneous NETosis after 4 hours in culture (B), and patients have elevated circulating NETs as measured by MPO::dsDNA and NE::dsDNA complexes (C). D) COPA patient-derived basal epithelial cells fail to curb IFNB1 and CXCL10 transcript production in response to dsDNA transfection. Values are mean +/- SEM. Statistical significance was determined using Two-Way ANOVA. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
To cite this abstract in AMA style:
Moore R, Huo L, Koziel C, Stripp B, Vogel T, Wallace D, Jefferies C. Mutant COPA Permits Aberrant NETosis and Exacerbates Autoimmune Lung Disease [abstract]. Arthritis Rheumatol. 2025; 77 (suppl 9). https://acrabstracts.org/abstract/mutant-copa-permits-aberrant-netosis-and-exacerbates-autoimmune-lung-disease/. Accessed .« Back to ACR Convergence 2025
ACR Meeting Abstracts - https://acrabstracts.org/abstract/mutant-copa-permits-aberrant-netosis-and-exacerbates-autoimmune-lung-disease/