Session Information
Date: Monday, November 8, 2021
Session Type: Poster Session C
Session Time: 8:30AM-10:30AM
Background/Purpose: Pulmonary Arterial Hypertension (PAH) is a rare but potentially fatal complication of Adult-Onset Still’s Disease (AOSD). To date, only isolated observations have been published. Our objective was to establish the largest case series of AOSD patients with PAH, and to describe their clinical profile, evolution and response to treatments.
Methods: Cases were retrospectively identified from the French PAH network database and from an online call of the “Club Rhumatismes et Inflammation” (http://www.cri-net.com). To be included, all patients had to fulfil the Yamaguchi or Fautrel criteria for AOSD and PAH had to be confirmed by right heart catheterization. The data were collected using a standardized questionnaire.
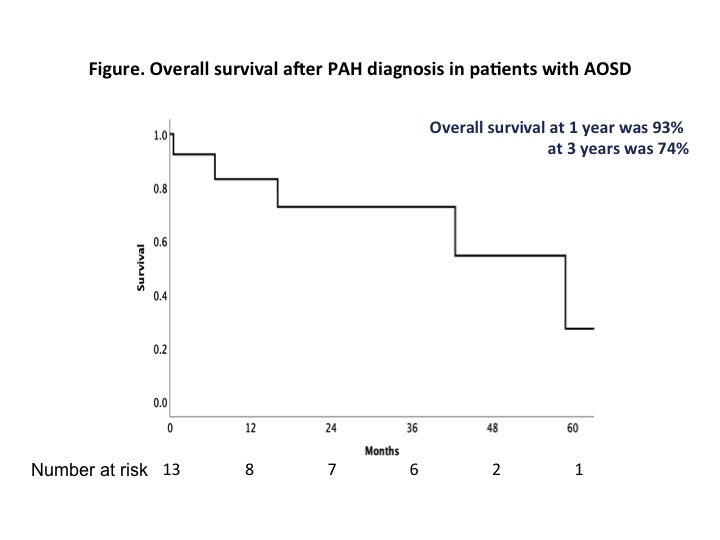
Results: Thirteen patients were identified. All cases of PAH occured after the diagnosis of AOSD was made. All were female, the mean age at PAH diagnosis was 32± 12 years, 2 (15%) patients were Caucasian, 6 (46%) from Sub-Saharan Africa, 1 (8%) from Asia and 4 (31%) from West Indies. Only 2 (15%) patients were smokers. All patients had a systemic onset of AOSD, 12 had a polycyclic and 1 a chronic articular evolution, and the mean delay between AOSD and PAH diagnosis was 2.9 (range 1.7 -5.4) years. PAH diagnosis was concomitant of AOSD flare in 11 (85%) patients. At PAH diagnosis, patients were receiving the following treatments: 13 (100%) corticosteroids (median dose 12 mg [interquartile range (IQR) 10-20]), 4 (31%) methotrexate, 9 (69%) interleukin (IL)-1 inhibitors (exposure median duration 7.1 months [IQR 4.4-10.1]), none IL-6 inhibitors, 2 (15%) TNF inhibitors. PAH was severe at diagnosis: 2 (15%), 7 (54%) and 4 (31%) patients were in NYHA functional class II, III and IV, respectively, with a median 6-minute walk distance of 289 m [IQR 0-448], a mean pulmonary arterial pressure of 41 ± 12 mmHg, a mean pulmonary arterial occlusion pressure of 6 ± 3 mmHg, a mean cardiac output of 3.9 ± 1.2 L/min, a mean cardiac index of 2.5 ± 0.9 L/min/m2 and a median pulmonary vascular resistance of 7 Wood Units [IQR 6-11]. The treatment prescribed after PAH diagnosis is detailed in the table. The median follow-up was 34 months [IQR 10-42]. Five patients (38.5 %) died. The haemodynamic response to PAH treatment seemed to be dissociated from the prognosis since three patients died while their haemodynamic had improved or almost normalized. Three patients died during an AOSD flare with disseminated intravascular coagulation. Figure 1 shows the overall survival which was 93% at 1 year and 74% at 3 years.
Conclusion: PAH is a rare but potentially severe complication of AOSD. It affected exclusively women in our case series. AOSD remission should be physicians’ objective, since PAH seems to occur when the underlying disease is not controlled.

To cite this abstract in AMA style:
Mitrovic S, Boucly a, Lazaro E, SCHLEINITZ N, Bloch-Queyrat C, Christides C, Pouchot J, humbert m, Montani D, savale l, Jaïs X, Sitbon O, Fautrel B. Pulmonary Arterial Hypertension in Adult-Onset Still’s Disease: A Case Series of 13 Patients [abstract]. Arthritis Rheumatol. 2021; 73 (suppl 9). https://acrabstracts.org/abstract/pulmonary-arterial-hypertension-in-adult-onset-stills-disease-a-case-series-of-13-patients/. Accessed .« Back to ACR Convergence 2021
ACR Meeting Abstracts - https://acrabstracts.org/abstract/pulmonary-arterial-hypertension-in-adult-onset-stills-disease-a-case-series-of-13-patients/