Session Information
Date: Monday, October 22, 2018
Title: Systemic Sclerosis and Related Disorders – Basic Science Poster II
Session Type: ACR Poster Session B
Session Time: 9:00AM-11:00AM
Background/Purpose: Internal organ involvement is the primary cause of morbidity and mortality in systemic sclerosis (SSc). Here we tested the hypothesis generated from a meta-analysis of ten different SSc datasets, that any single patient with SSc would have the same deregulated molecular signatures across multiple organ systems, consistent with the systemic nature of the disease.
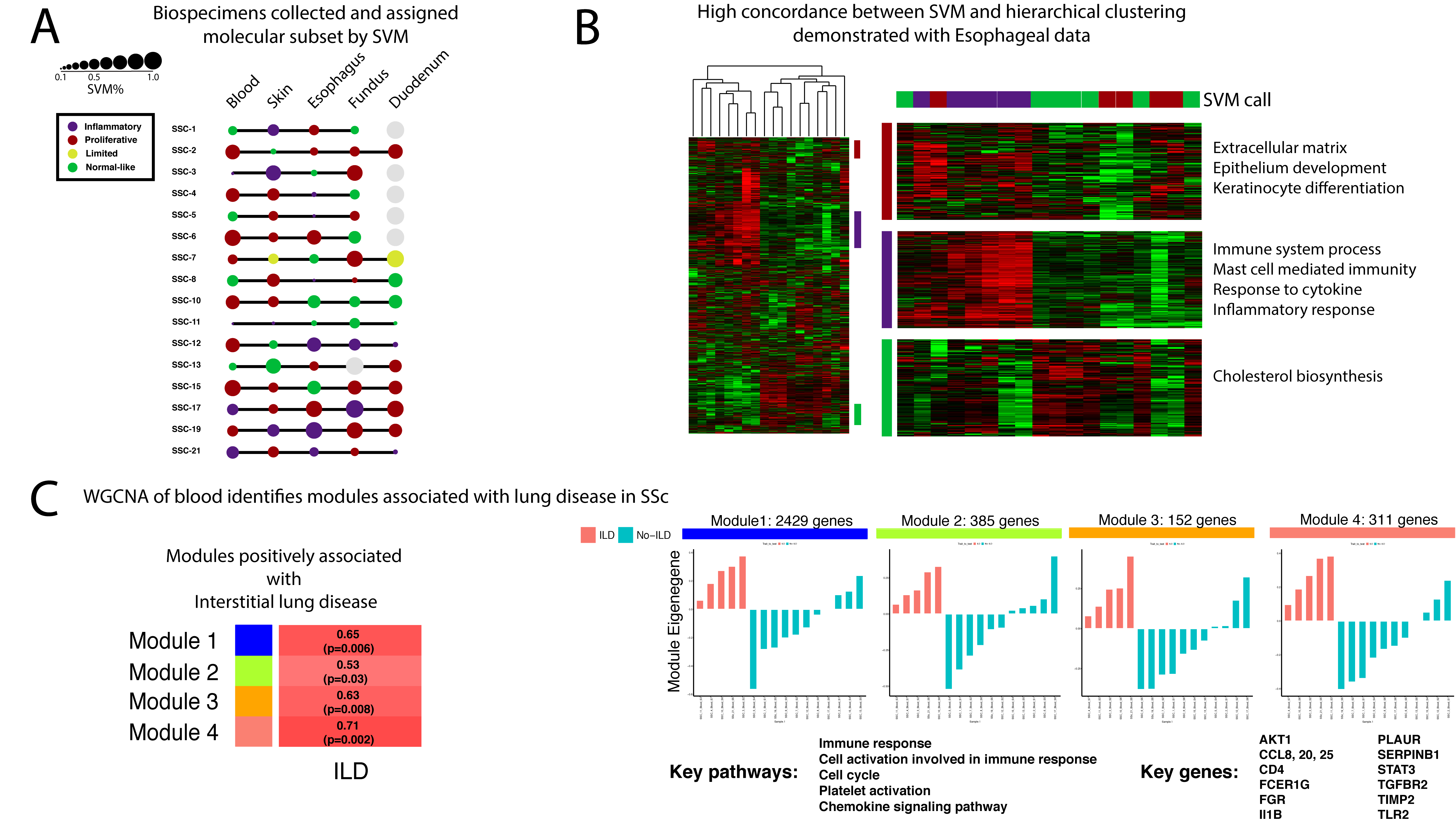
Methods: RNA sequencing (RNA-seq) was performed on at least four organ biospecimens (skin, esophagus, fundus, duodenum, and blood) from 16 patients who met 2013 ACR/EULAR criteria for SSc. RNA was sequenced by 75bp paired-end RNA-seq at >80 million reads per sample and aligned to the reference genome (hg19). DEseq2 was used to identify differentially expressed genes across the various tissue pairs. Each sample was assigned to an intrinsic gene expression subset (inflammatory, proliferative, limited, or normal-like) using a Support Vector Machine (SVM) classifier and unsupervised hierarchical clustering with profiles of normalized Reads Per Kilobase of transcript per Million mapped reads (RPKM) values. Weighted Gene Co-Expression Analysis (WGCNA) was used to identify modules across tissues.
Results: Hierarchical clustering shows that biospecimens from the same tissue type cluster together regardless of whether a patient has SSc. We normalized each tissue specific dataset to allow comparative analyses of SSc biology across the tissues. We recapitulated the intrinsic subsets previously identified in the skin and esophagus of SSc patients. SVM classification further identified intrinsic subsets in the duodenum, fundus, and blood of patients with SSc. The majority of our patients (9/16) exhibit >60% concordance of intrinsic subset assignment across tissues. Importantly, all five organs were represented in the SVM calls of inflammatory, proliferative, and normal-like intrinsic subsets. WGCNA identifies modules of genes in blood of patients with SSc that are significantly associated with clinical variables. Most notably, four modules positively associated with interstitial lung disease in patients contain genes (STAT3, Il1B, TGFBR2) and are enriched in pathways (Immune response, cell cycle, chemokine signaling) that have been implicated in SSc pathogenesis.
Conclusion: We have completed collection of >4 biospecimens from 16 individuals with SSc. Our data shows that the intrinsic gene expression subsets, first identified in skin, exist systemically in patients with SSc. These molecular profiles are more consistent than chance within single individuals and are thus, a common feature in end-organ pathology in SSc. We have identified modules of genes in easily accessible tissues that can be used to assess interstitial lung disease (ILD) in patients. Modules associated with additional clinical variables (antibody status, esophageal pathology, and treatment) are currently being evaluated.
To cite this abstract in AMA style:
Mehta BK, Franks J, Wang Y, Cai G, Toledo DM, Wood TA, Archambault KA, Kosarek N, Kolstad KD, Stark M, Valenzuela A, Fiorentino D, Fernandez-Becker N, Becker L, Nguyen L, Clarke J, Boin F, Wolters P, Chung L, Whitfield ML. Multi-Organ RNA-Sequencing of Patients with Systemic Sclerosis (SSc) Finds That Intrinsic Subsets Are Conserved across Organ Systems [abstract]. Arthritis Rheumatol. 2018; 70 (suppl 9). https://acrabstracts.org/abstract/multi-organ-rna-sequencing-of-patients-with-systemic-sclerosis-ssc-finds-that-intrinsic-subsets-are-conserved-across-organ-systems-2/. Accessed .« Back to 2018 ACR/ARHP Annual Meeting
ACR Meeting Abstracts - https://acrabstracts.org/abstract/multi-organ-rna-sequencing-of-patients-with-systemic-sclerosis-ssc-finds-that-intrinsic-subsets-are-conserved-across-organ-systems-2/