Session Information
Date: Sunday, October 21, 2018
Title: Systemic Sclerosis and Related Disorders – Basic Science Poster I
Session Type: ACR Poster Session A
Session Time: 9:00AM-11:00AM
Background/Purpose: Though transforming growth factor-b (TGF-b) plays a fundamental role in the pathogenesis of SSc, the mechanism by which TGF-b signaling acts in SSc remains largely unclear. The aim of this study is to clarify the underlying mechanism of TGF-b signaling over-activation in SSc, especially epigenetic manners.
Methods: Twenty SSc patients and twenty-one age, gender-matched healthy individuals were enrolled in this study. Luciferase analysis was used to evaluate the binding capacity of miRNA-3606-3p to TGFBR2. Western blot and qPCR were used to detect the expression of miRNA, TGFBR2, p-SMAD2/3 and collagen.
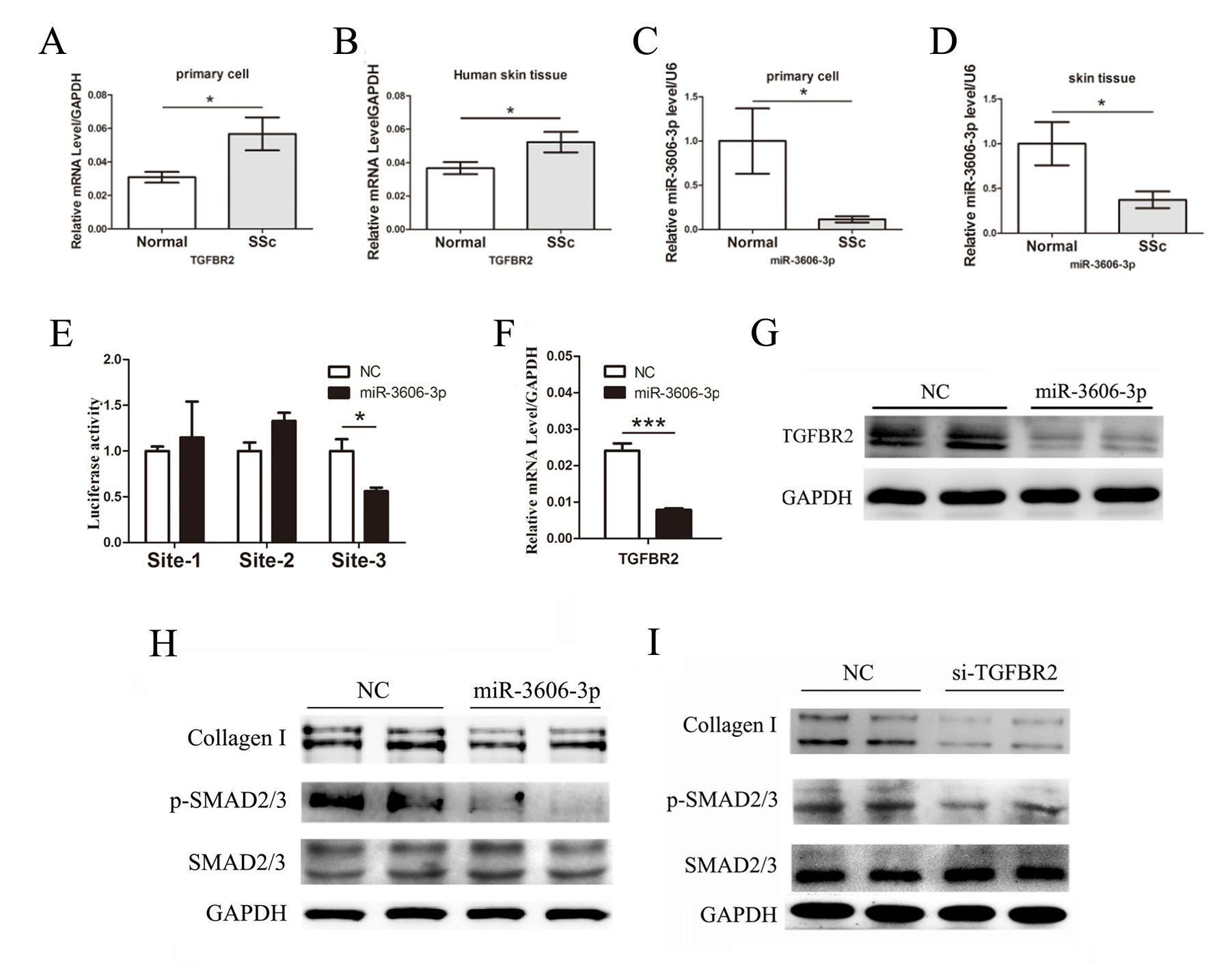
Results: Transcription level of TGFBR2 was increased in both SSc skin samples and fibroblasts (Fig. 1A and 1B). In contrast, miR-3606-3p, a predicted miRNA of TGFBR2, was downregulated in SSc fibroblasts and skin tissues (Fig. 1C and 1D).
Based on the above findings, we then determined whether TGFBR2 is the direct target for miR-3606-3p. Three TGFBR2 3′-UTR wild type-containing binding sites (site 1, site 2 and site 3) of miR-3606-3p were subcloned into the pmirGLO vector, and co-transfected with a luciferase construct containing miR-3606-3p mimics into primary dermal fibroblast cells. As illustrated in Fig. 1E, the relative luciferase activity was only significantly decreased in the TGFBR2 3′-UTR wild type site 3 group, whereas there was no significant difference in the TGFBR2 3′-UTR wild type site 1 or site 2 group, suggesting that miR-3606-3p may selectively bind to the sequence containing site 3 of the TGFBR2 3′-UTR. Furthermore, overexpression of miR-3606-3p significantly diminished the level of TGFBR2 mRNA (Fig. 1F) and TGFBR2 protein (Fig. 1G).
Next, we examined whether the repression of TGFBR2 mediated by miR-3606-3p would disrupt TGF-b/Smad signaling in fibroblasts. Western blot analysis showed that the transfection of primary fibroblast cells with miR-3606-3p mimics significantly reduced the protein levels of type I collagen and p-SMAD2/3 (Fig. 1H). We then analyzed the expression of type I collagen and p-SMAD2/3 in response to TGFBR2 downregulation in the fibroblasts. Interestingly, siRNA-induced TGFBR2 downregulation revealed similar results to those obtained via miR-3606-3p overexpression in the fibroblasts (Fig. 1I).
Conclusion: Our findings demonstrated that increased TGFBR2 could be responsible for the hyperactive TGF-b signaling observed in SSc. We identified a pivotal role for miR-3606-3p in SSc, which inhibits TGF-b signaling, at least partly, through TGFBR2 repression. The results suggest that the regulation of miR-3606-3p/TGFBR2 could be a promising therapeutic strategy for treatment of fibrosis.
Figure 1
To cite this abstract in AMA style:
Shi X, Liu Q, Tu W, Mei X, Jin L, Wu W, Wang J. MiR-3606-3p Inhibits Systemic Sclerosis through Targeting TGF-β Receptor II [abstract]. Arthritis Rheumatol. 2018; 70 (suppl 9). https://acrabstracts.org/abstract/mir-3606-3p-inhibits-systemic-sclerosis-through-targeting-tgf-%ce%b2-receptor-ii/. Accessed .« Back to 2018 ACR/ARHP Annual Meeting
ACR Meeting Abstracts - https://acrabstracts.org/abstract/mir-3606-3p-inhibits-systemic-sclerosis-through-targeting-tgf-%ce%b2-receptor-ii/