Session Information
Session Type: ACR Poster Session C
Session Time: 9:00AM-11:00AM
Background/Purpose: Glucocorticoids (GC) remain the frontline treatment in Systemic Lupus Erythematosus (SLE) despite their predictable metabolic adverse effects. Type I interferons (IFN), produced by TLR7 and TLR9 activated plasmacytoid dendritic cells (pDC), play a critical role in SLE pathogenesis, but are not suppressed by GC. Glucocorticoid-Induced Leucine Zipper (GILZ) is an endogenous anti-inflammatory protein, which regulates T and B cell activation and is suppressed in SLE; whether GILZ regulates type 1 IFN production in SLE is not known. We tested the hypothesis that GILZ inhibits Type I IFN production in SLE.
Methods: We performed in vitro analysis on pDC and bone marrow-derived DC (BMDC), and in vivo studies, of WT and GILZ-/- mice using stimuli of TLR4 (LPS), TLR7 (Imiquimod), TLR7/8 (Resiquimod) and TLR9 (CpG). IFN was measured using a IFN luciferase assay, ELISA and Luminex. IFN-stimulated gene signatures (ISG) were measured by qPCR. To determine whether GILZ regulates IFN in human SLE, we mined a public gene expression dataset GSE61635. We examined IRF7 and NF-κB involvement in GILZ effects using ChIP and reporter assays.
Results: GILZ deficiency resulted in increased pDC and BMDC secretion of IFN in response to TLR7, TLR7/8 and TLR9 stimulation. GILZ deficiency was also associated with increased ISG in naïve spleen cells, and TLR-stimulated pDC (Fig 1A) and BMDC of GILZ-/- mice. Correspondingly, increased IFN was seen in GILZ-/- mice in response to TLR7/8 stimulation in vivo. In human SLE whole blood, GILZ mRNA was negatively correlated with ISG (IFI44, IFI44L, RSAD2, IFI27) (Fig 1B). In BMDC, TLR activation suppressed GILZ, but increased IRF7 expression. CHIP-qPCR showed GILZ enrichment at the IRF7 locus in unstimulated cells, which decreased following TLR7/8/9 activation, indicating direct binding of GILZ to the IRF7 locus as the mechanism through which it regulates IFNα production. In addition, TLR7/8/9-induced NF-κB activation was suppressed by GILZ, and increased IFNβ in GILZ-/- BMDC was inhibited by NF-κB inhibition.
Conclusion: GILZ is a novel endogenous regulator of Type I IFN production in vitro, in vivo, and in human SLE, which has direct effects on both IRF7 and NF-κB and thereby both IFNα and IFNβ. This suggests that suppressed GILZ is permissive for IFN activation in SLE, and represents the mechanism through which IFN escapes glucocorticoid suppression.
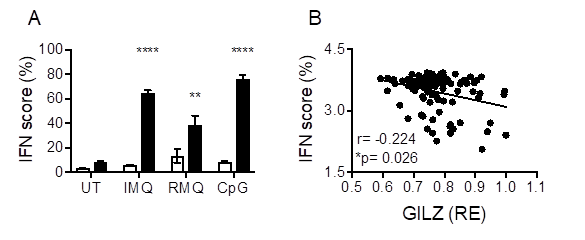
To cite this abstract in AMA style:
Nataraja C, Morand E, Lee J, Bennett T, Flynn J, Harris J, Jones S. Glucocorticoid-Induced Leucine Zipper (GILZ) Is a Novel Checkpoint Regulator of Type I Interferon (IFN) Production in SLE [abstract]. Arthritis Rheumatol. 2018; 70 (suppl 9). https://acrabstracts.org/abstract/glucocorticoid-induced-leucine-zipper-gilz-is-a-novel-checkpoint-regulator-of-type-i-interferon-ifn-production-in-sle/. Accessed .« Back to 2018 ACR/ARHP Annual Meeting
ACR Meeting Abstracts - https://acrabstracts.org/abstract/glucocorticoid-induced-leucine-zipper-gilz-is-a-novel-checkpoint-regulator-of-type-i-interferon-ifn-production-in-sle/