Session Information
Title: Systemic Sclerosis, Fibrosing Syndromes and Raynaud’s – Pathogenesis, Animal Models and Genetics
Session Type: Abstract Submissions (ACR)
Background/Purpose:
Scleroderma, defined as pathologic fibrosis of the skin, has many clinical presentations. In the most commonly recognized form, systemic sclerosis (SSc), previously healthy individuals acquire fibrosis of the skin and viscera associated with autoantibodies. Although a genetic contribution to SSc has been established, familial recurrence is rare and causal genes have not been found. The study of SSc is hindered by a lack of animal models that faithfully recapitulate this complex disease. In contrast, a rare disorder, stiff skin syndrome (SSS), shows childhood onset of diffuse skin fibrosis with autosomal dominant inheritance and no visceral fibrosis or autoimmunity. Both disorders have been linked to TGFβ. SSS is caused by heterozygous missense mutations in the gene encoding fibrillin-1, a major constituent of extracellular microfibrils. SSS mutations localize to the only domain that harbors an Arg-Gly-Asp motif that mediates cell-matrix interaction by integrin-binding.
Methods:
Two SSS mouse models were generated by homologous recombination and either bred to Itgb3+/- mice or treated with antibodies by IP injection. Primary human dermal fibroblast cultures were derived from forearm biopsies and stimulated with 2 ng/mL TGFβ1. Analyses included skin stiffness scoring, histology, flow cytometry, Western and Northern blotting, and qPCR. Data are shown as standard boxplots (R statistical software) and analyzed with 2-tailed t tests.
Results:
Knock-in mutations in Fbn1 in mice that either reproduce a naturally-occurring SSS mutation (Fbn1W1572C/+) or impose an obligate loss of integrin binding (Fbn1D1545E/+), are sufficient to initiate and maintain dermal fibrosis. This associates with upregulation of surface expression of β1 and αvβ3 integrins by dermal cells. Treatment with β1 integrin activating (β1aAb) for 12 weeks reduces active αvβ3 in the dermis and prevents stiffness and skin fibrosis (Fig.1). Notably, genetic ablation of integrin β3 also prevents the phenotype and TGFβ antagonism, commenced at a later age, completely reverses it. In vitro, SSc patient cells also show aberrant integrin expression and function that contributes to a fundamental change in the signaling properties of ligand-activated TGFβ receptors, favoring activation of extracellular-regulated kinase (ERK). Integrin- or ERK-modulating treatments normalized the pro-fibrotic repertoire in SSc cells, increasing levels of matrix-inhibiting microRNA-29, and reducing both ERK activation and collagen expression (Fig.2).
Conclusion:
This work demonstrates the potential to reverse established dermal fibrosis in a model of human disease and shows that despite the phenotypic differences between SSS and SSc, study of SSS animal models has offered a pathogenic sequence for scleroderma that suggests therapies for SSc, including β1 integrin activation and blockade of β3 integrin, TGFβ or ERK signaling.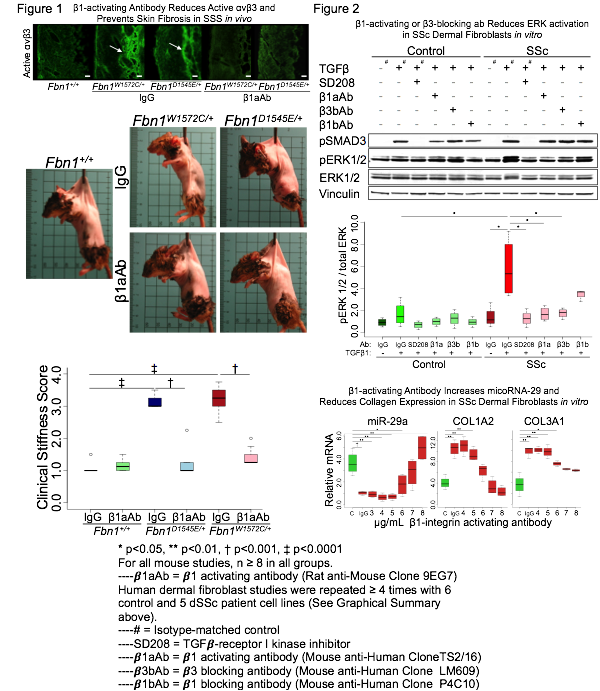
Disclosure:
E. E. Gerber,
None;
F. M. Wigley,
None;
E. C. Davis,
None;
D. L. Huso,
None;
H. C. Dietz,
None.
« Back to 2012 ACR/ARHP Annual Meeting
ACR Meeting Abstracts - https://acrabstracts.org/abstract/crosstalk-between-integrins-and-tgf%ce%b2-in-the-pathogenesis-and-treatment-of-multiple-presentations-of-scleroderma/