Session Information
Session Type: Poster Session A
Session Time: 10:30AM-12:30PM
Background/Purpose: Multiple genomic regions are known to confer risk for JIA. However, identifying the SNPs that exert the biological effects that confer risk, and therefore the target genes, is confounded by linkage disequilibrium (LD). LD makes the true risk-driving SNPs statistically indistinguishable from those (the majority) with which they are co-inherited. We used novel analytics and wet lab procedures to identify three SNPs within JIA risk regions whose biological properties make them likely disease-driving variants.
Methods: Our groups have developed independent methods for assessing the likelihood that a SNP on a disease haplotype exerts risk-driving biological effects. The first is an AI-based approach that captures the effect of JIA SNPs on each of the distinct stages of enhancer assembly (as we have previously shown that JIA risk variants are likely to be situated in active or poised enhancers). We trained sequence-to-activity models to predict three different types of genomic data, ATAC-seq, PRO-cap, and PRO-seq, that capture different stages of enhancer assembly. The second method predicts allelic effects on DNA shape/topology, a critical feature of DNA function (e.g., transcription factor binding). To verify the utility of these approaches, we undertook wet lab confirmation of computational predictions using enhancer reporter and DNA pulldown assays.
Results: Using the AI-based method for assessing enhancer (and transcriptional) effects, we identified a SNP within the JIA-associated IRF1 locus, rs2548998 G to A, as likely to influence transcription. This SNP lies within a large enhancer cluster upstream of the IRF1 transcription start site, marked by both H3K27ac ChIPseq and PROseq-dREG peaks. Using a luciferase enhancer reporter assay, we demonstrated that the rs2548998 G®A allele showed 2x more activity than the common allele in activated primary human CD4+ T cells (Figure 1). We used a DNA pulldown assay to corroborate predictions from DNA topology analyses regarding effects of SNPs’ effects on transcription factor binding. We tested 2 SNPs, rs4147359 G to A, within an a CREB1 binding site upstream of IL2RA (using nuclei extracted from Jurkat T cells), and rs7234029 A®G, situated in a GATA1 binding site within the first intron of PTPN2 (using myeloid K562 cells; GATA1 is not expressed in lymphoid cells). Figure 2 summarizes the results of these experiments. The SNP, rs4147359 G®A reduced CREB1 by approximately 2-fold compared to the common allele (p< 0.01), while rs7234029 A®G reduced GATA1 binding by more than 4-fold compared to the common allele (p< 0.05).
Conclusion: We demonstrate that, by using a combination of computational techniques that predict different features of DNA/chromatin, and corroborating computational predictions with wet lab techniques, we can identify true risk-driving alleles on JIA risk haplotypes.
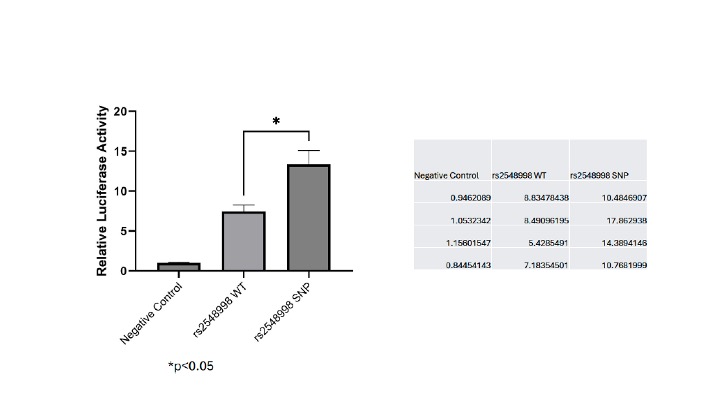 Figure 1 – Results of an enhancer reporter assay
Figure 1 – Results of an enhancer reporter assay
performed in CD3/CD28/IL2-activated primary humanCD4+ T cells. We compared the common allele
(“rs2548998 WT”) with the G to A variant (“rs2548998 SNP”). The SNP showed 2-fold enhancer activity compared with the common allele. Raw data from each of 4 experiments (relative luciferase expression) is shown in the table on the right. The negative control consisted of the green fluorescence protein vector without reporter vector without the enhancer construct inserted.
.jpg) Figure 2 – Identification of proteins with different binding affinity to common allele or allele of single nucleotide polymorphismsidentified via DNA topology analyses as likely to have significant effects on transcription factor binding. (A) Western blot verification of DNA pull-down assay. Nuclear protein extracts from Jurkat cells were incubated with Streptavidin MicroBeads and biotin-labeled probes representing the common allele or allele of rs4147359 (GA, TF CREB1). Nuclear protein extracts from K562 cells were incubated with Streptavidin MicroBeads and biotin-labeled probes representing the common allele or allele of rs7234029 (AG, TF
Figure 2 – Identification of proteins with different binding affinity to common allele or allele of single nucleotide polymorphismsidentified via DNA topology analyses as likely to have significant effects on transcription factor binding. (A) Western blot verification of DNA pull-down assay. Nuclear protein extracts from Jurkat cells were incubated with Streptavidin MicroBeads and biotin-labeled probes representing the common allele or allele of rs4147359 (GA, TF CREB1). Nuclear protein extracts from K562 cells were incubated with Streptavidin MicroBeads and biotin-labeled probes representing the common allele or allele of rs7234029 (AG, TF
GATA1). (B) The protein levels were quantified using the iBright Analysis System (Thermo Fisher Scientific). Significant differences were marked with an asterisk (*p < 0.05; **p < 0.01; n=4).
To cite this abstract in AMA style:
He A, Ainsworth H, Jiang K, Khtovatkova E, Chen Y, Langefeld C, Danko C, Jarvis J. Computational and Laboratory Identification of Risk-Driving Alleles on Juvenile Idiopathic Arthritis (JIA)-Associated Haplotypes [abstract]. Arthritis Rheumatol. 2025; 77 (suppl 9). https://acrabstracts.org/abstract/computational-and-laboratory-identification-of-risk-driving-alleles-on-juvenile-idiopathic-arthritis-jia-associated-haplotypes/. Accessed .« Back to ACR Convergence 2025
ACR Meeting Abstracts - https://acrabstracts.org/abstract/computational-and-laboratory-identification-of-risk-driving-alleles-on-juvenile-idiopathic-arthritis-jia-associated-haplotypes/