Session Information
Date: Tuesday, November 7, 2017
Title: Rheumatoid Arthritis – Small Molecules, Biologics and Gene Therapy III: Biosimilars Therapy
Session Type: ACR Concurrent Abstract Session
Session Time: 2:30PM-4:00PM
Background/Purpose: GP2013, is a biosimilar to European Union (EU) approved reference rituximab (RTX), developed in a stepwise approach that adheres to stringent biosimilar development guidelines. The biosimilarity of GP2013 and RTX has been confirmed preclinically and in patients with RA and follicular lymphoma. Switching patients from a reference product to its biosimilar has to be shown for regulatory approval in some regions. In this study, the safety and immunogenicity of the switch from RTX to GP2013 was compared with the continued RTX in patients with active RA.
Methods: This randomized, double-blind, parallel-group study in adult patients with active RA was conducted across the United States (US; 28 centers) and the EU (26 centers), who had received the last treatment with RTX 6-18 months prior to randomization and required RTX re‑treatment. Patients were randomized (1:1) to receive 1000 mg intravenous infusions of either GP2013 (switch group) or continue RTX (control group) on days 1 and 15. Methotrexate (7.5-25 mg/week) and folic acid were given in the same stable dose as before randomization. Safety assessments included the incidence of hypersensitivity; infusion-related and anaphylactic reactions; immunogenicity (antidrug antibody [ADA] development); the incidence of adverse events (AE), and other safety parameters (vital signs and laboratory parameters). Patients were followed up for 24 weeks.
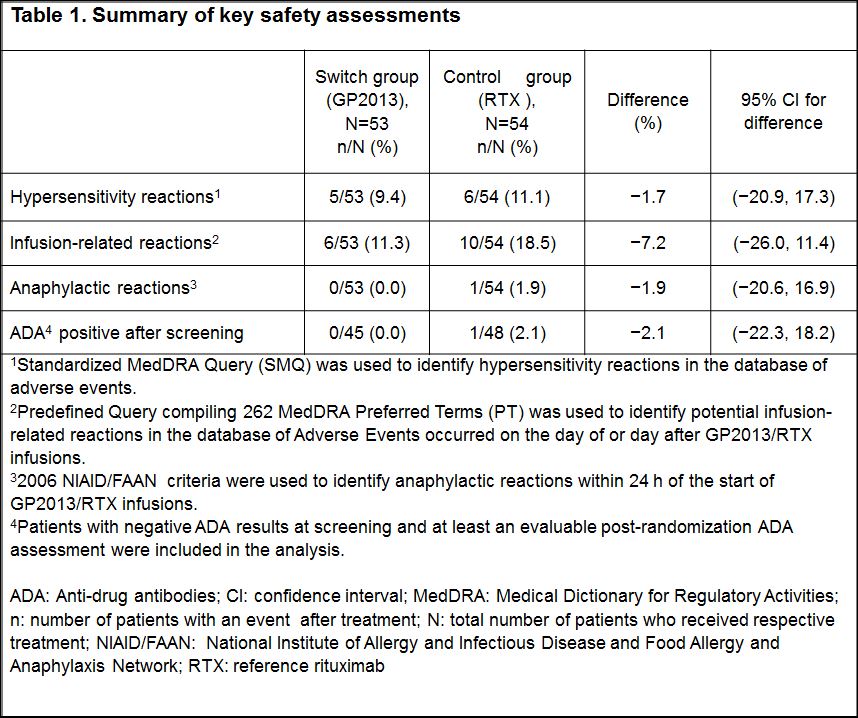
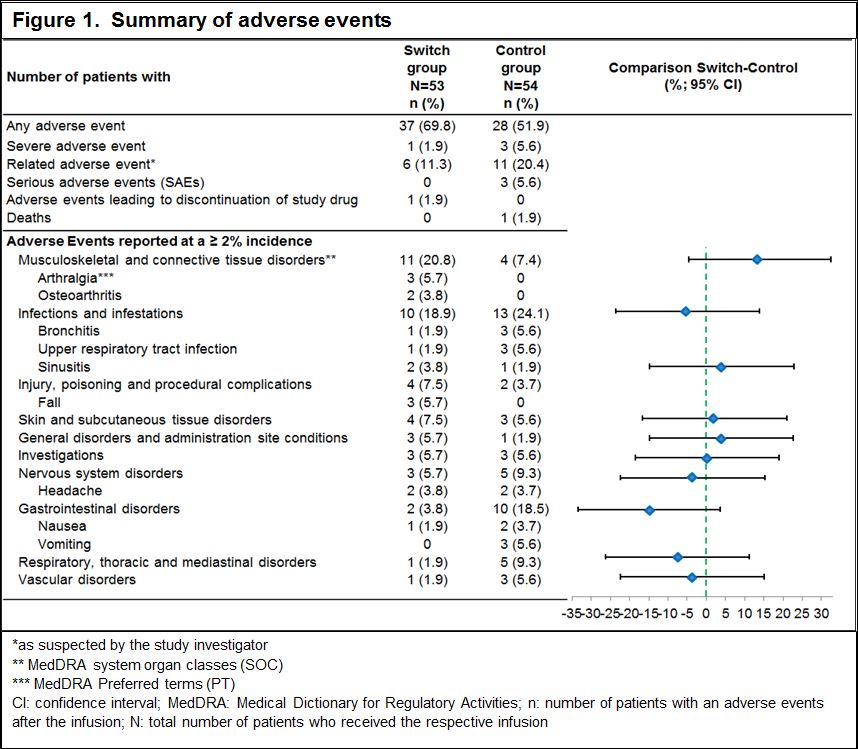
Results: Of the 107 patients randomized to the switch group (n=53) or control group (n=54), majority of patients completed the study up to 24 weeks (94.3% vs 96.3%). Demographic and baseline characteristics were well balanced between the groups. The incidence of hypersensitivity (9.4% vs 11.1%) and infusion-related reactions (11.3% vs 18.5%) were low and similar in both groups. In the control group, 1 patient developed an anaphylactic reaction within 24 h of infusion. Among the patients, who tested negative for ADA at screening, only 1 patient in the control group tested positive for ADAs in all visits after the first infusion (Table 1). No patient had ADA of neutralizing capacity. The rate of AEs was similar between the groups, serious AE occurred only in the control group (Figure 1). Although more patients in the switch group had AEs in some AE categories, these differences could not be attributed to a cluster of specific events. The majority of patients in both treatment groups did not have newly occurring laboratory abnormalities.
Conclusion: The safety profile, in particular hypersensitivity reactions and occurrence of ADAs, of patients who switched from RTX to GP2013 was comparable with the patients who received continued treatment with RTX.
To cite this abstract in AMA style:
Tony HP, Schulze-Koops H, Krüger K, Cohen SB, Kivitz AJ, Jeka S, Vereckei E, Cen L, Kollins D. Comparison of Switching from the Originator Rituximab to the Biosimilar Rituximab GP2013 or Re‑Treatment with the Originator Rituximab in Patients with Active Rheumatoid Arthritis: Safety and Immunogenicity Results from a Multicenter, Randomized, Double-Blind Study [abstract]. Arthritis Rheumatol. 2017; 69 (suppl 10). https://acrabstracts.org/abstract/comparison-of-switching-from-the-originator-rituximab-to-the-biosimilar-rituximab-gp2013-or-re%e2%80%91treatment-with-the-originator-rituximab-in-patients-with-active-rheumatoid-arthritis-safety-and/. Accessed .« Back to 2017 ACR/ARHP Annual Meeting
ACR Meeting Abstracts - https://acrabstracts.org/abstract/comparison-of-switching-from-the-originator-rituximab-to-the-biosimilar-rituximab-gp2013-or-re%e2%80%91treatment-with-the-originator-rituximab-in-patients-with-active-rheumatoid-arthritis-safety-and/