Session Information
Date: Monday, November 8, 2021
Session Type: Abstract Session
Session Time: 4:45PM-5:00PM
Background/Purpose: VEXAS (vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic) syndrome is a recently described myeloid-driven autoinflammatory condition caused by somatic mutations affecting methionine-41 (p.Met41) in the UBA1 gene which encodes the major E1 enzyme which initiates ubiquitylation.1 The purpose of this study was to describe a series of cases highlighting the clinical features and outcomes of patients with this condition.
Methods: The divisions of rheumatology and hematology at Mayo Clinic, Rochester, Minnesota and the rheumatology division of the University of California, Los Angeles, (UCLA) identified all patients who underwent testing for UBA1 somatic mutations. The National Institutes of Health (NIH) performed testing for somatic mutations of the UBA1 gene, using methods previously described.1 No patient included in this study was part of the original NIH cohort.1 Demographic information, clinical characteristics, peripheral blood and bone marrow biopsy findings, treatment regimens and outcome were abstracted from direct medical chart review. Continuous data are presented as mean ± standard deviation (SD) or median [interquartile range (IQR) (25th percentile, 75th percentile)] and categorical variables as percentages.
Results: Nine men with somatic mutations in the UBA1 gene were identified. Clinical features are noted in Table 1. The most frequent variant was p.Met41Thr (7/9, 78%). The median age at VEXAS diagnosis was 74 (67, 76.5) years, and patients had a median duration of symptoms for 4 years prior to diagnosis. Refractory constitutional symptoms (88%), ear/nose chondritis (55%) and inflammatory arthritis (55%) were common clinical features. Vasculitis was noted in four patients (44%). All four patients had cutaneous vasculitis, but additionally one (#4) had evidence of renal peri-tubular capillaritis, one (#8) had type III cryoglobulinemia with digital gangrene and one (#9) had large-vessel vasculitis. All patients had significantly elevated inflammatory markers and macrocytic anemia. Thrombocytopenia was present in 66% at VEXAS diagnosis. Eight patients had bone marrow biopsies performed. All bone marrows were hypercellular and there was vacuolization of the erythroid (100%) and/or myeloid precursors (75%). Glucocorticoids attenuated symptoms at doses ≥ 20 mg/day but no other immunosuppressive agent showed consistent long-term disease control. One patient with co-existing plasma cell myeloma received plasma cell directed therapy (bortezomib, lenalidomide, dexamethasone induction therapy) followed by autologous stem cell transplant resulting in improvement of the inflammatory response, which is a novel finding (Figure 1).
Conclusion: VEXAS syndrome is a clinically heterogenous, treatment-refractory inflammatory condition caused by somatic mutation of the UBA1 gene. Patients present with overlapping rheumatologic manifestations and persistent hematologic abnormalities. Optimal treatment regimens remain to be defined. The utility of bone marrow transplantation requires further investigation.
References: 1Beck DB, Ferrada KA, Sikora KA, et al. Somatic Mutations in UBA1 and Severe Adult-Onset Autoinflammatory Disease. NEJM 220;383:2628-28.
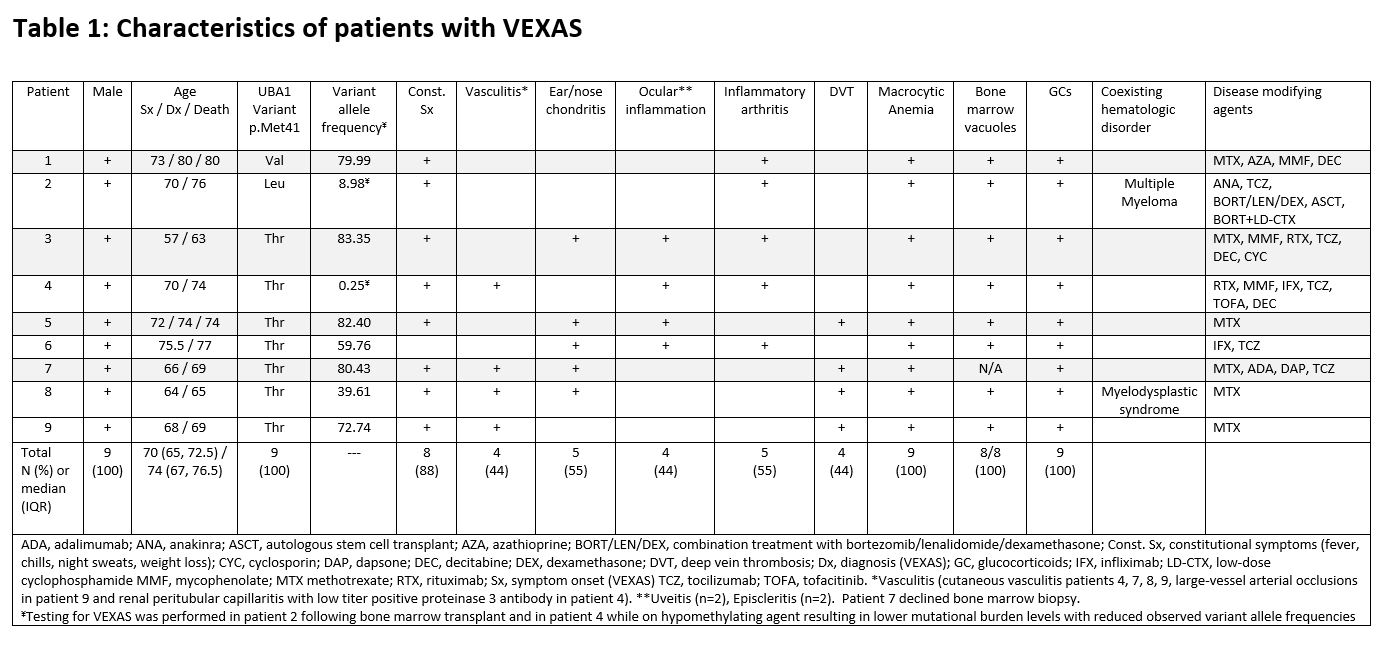 Table 1: Clinical Characteristics of Patients with VEXAS
Table 1: Clinical Characteristics of Patients with VEXAS
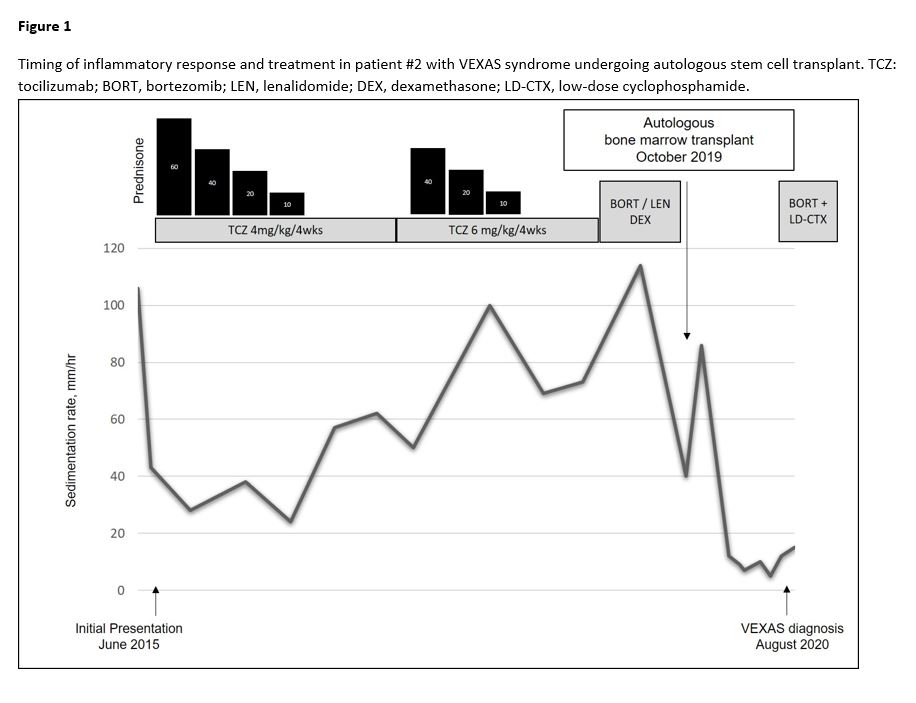 Pre and Post transplant inflammatory response in patient #2
Pre and Post transplant inflammatory response in patient #2
To cite this abstract in AMA style:
Koster M, Kourelis T, Reichard K, Kermani T, Beck D, Ospina Cardona D, Samec M, Mangaonkar A, Begna K, Hook C, Oliveira J, Nasr S, Tiong B, Patnaik M, Burke M, Michet C, Warrington K. Clinical Heterogeneity of the VEXAS Syndrome: A Case Series [abstract]. Arthritis Rheumatol. 2021; 73 (suppl 9). https://acrabstracts.org/abstract/clinical-heterogeneity-of-the-vexas-syndrome-a-case-series/. Accessed .« Back to ACR Convergence 2021
ACR Meeting Abstracts - https://acrabstracts.org/abstract/clinical-heterogeneity-of-the-vexas-syndrome-a-case-series/