Session Information
Date: Monday, October 22, 2018
Title: Systemic Sclerosis and Related Disorders – Basic Science Poster II
Session Type: ACR Poster Session B
Session Time: 9:00AM-11:00AM
Background/Purpose: Systemic Sclerosis (SSc) is an autoimmune disease characterized by fibrosis and inflammation. Multiple organ systems are affected including the skin, gastrointestinal tract, vasculature, and lungs(Katsumoto, Whitfield, & Connolly, 2011). It is a heterogeneous disease but gene expression analyses have defined four distinct molecular subtypes (inflammatory, fibroproliferative, normal-like, and limited). Prior studies have implicated microbial dysbiosis in SSc (Arron et al., 2014; Volkmann, 2017), but have not assessed the relationship between host immune processes/molecular subtype and microbial community characteristics. In this study we characterize the esophageal microbiome of patients with SSc and explore its relationship to host gene expression.
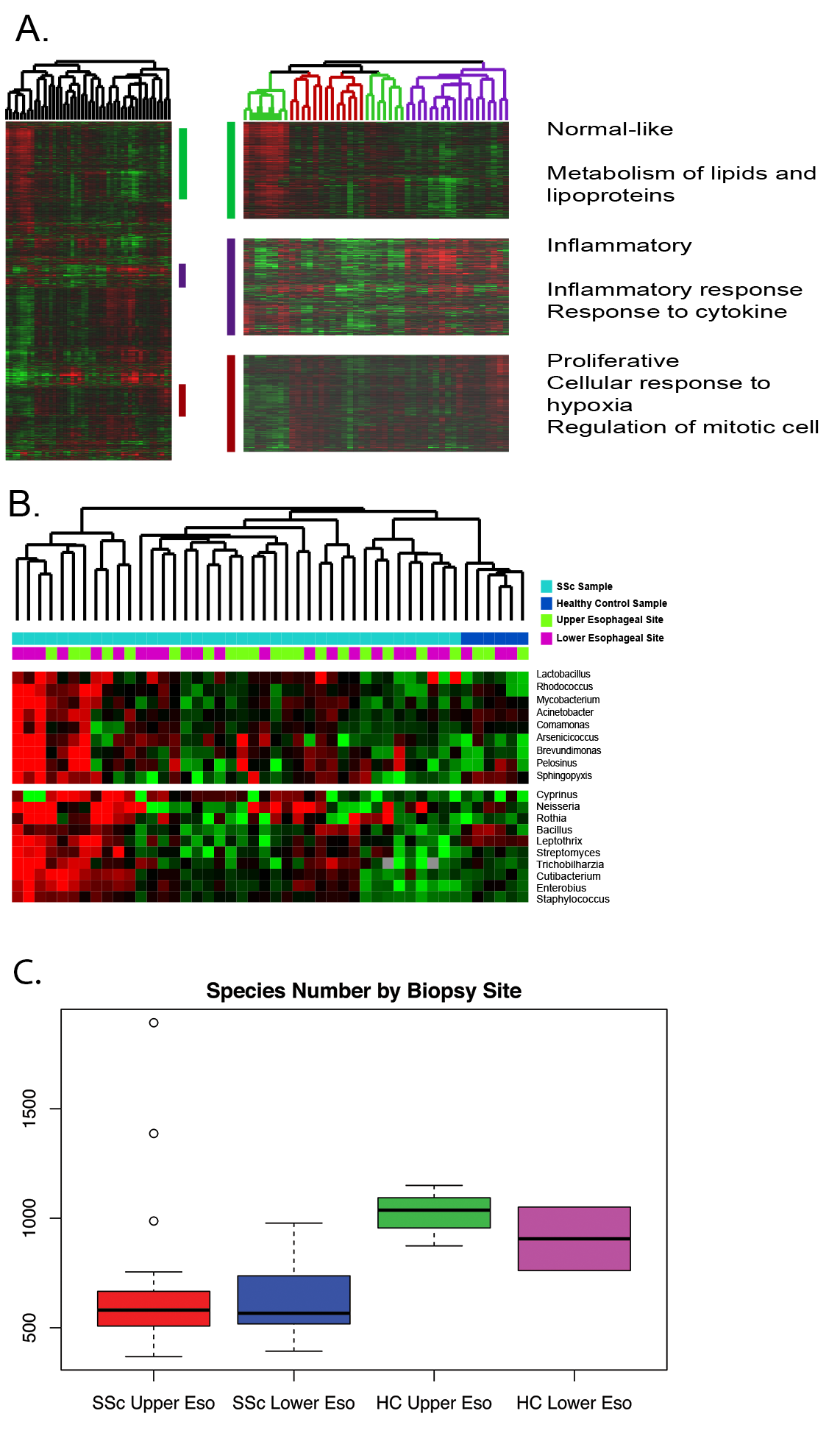
Methods: RNA-sequencing was performed on 19 patient and 4 healthy control (paired upper and lower) esophageal biopsies. Raw reads were aligned to hg19 via STAR and normalized to reads per kilobase million (RPKM). Intrinsic Gene Analysis (IGA) was performed to identify genes most similar between the upper and lower biopsies of a patient, but most dissimilar between patients, thus identifying genes that classify patients into SSc molecular subsets (inflammatory, proliferative, and normal-like). A 2% false discovery rate (FDR) was used, and genes passing this FDR were hierarchically clustered. Integrated Metagenomic Sequence Analysis (IMSA)(Dimon, Wood, Rabbitts, & Arron, 2013) was performed on sequencing reads from the same esophageal patient samples to extract microbial reads. Measures of species richness were compared between SSc patient samples across molecular subtypes and healthy controls.
Results: Consistent with prior data, hierarchical clustering of genes derived from IGA identified the molecular subsets of SSc (inflammatory, proliferative, and normal-like) in esophageal samples (Fig. 1A). IMSA identified more species in healthy control samples than SSc samples regardless of biopsy site (Fig. 1A). SSc patients and healthy controls differed in the abundance of Lactobacillus, Bacillus, and Rhodococcus. (Fig. 1B). These results are consistent with other work showing that dysbiosis of commensal bacteria is associated with SSc disease state (Volkmann et al, 2017)
Conclusion: SSc esophageal tissues recapitulate SSc processes and show an increase in abundance of potentially pathogenic commensal microbes when compared to healthy controls. Microbes do not hierarchically cluster samples by site, implicating a disease driven difference in microbial communities potentially also divided by SSc subtype.
To cite this abstract in AMA style:
Espinoza M, Mehta BK, Wang Y, Hoffmann A, Carns MA, Kosarek N, Wood TA, Hinchcliff M, Whitfield ML. Characterization of the Esophageal Microbiome in Patients with Systemic Sclerosis (SSc) [abstract]. Arthritis Rheumatol. 2018; 70 (suppl 9). https://acrabstracts.org/abstract/characterization-of-the-esophageal-microbiome-in-patients-with-systemic-sclerosis-ssc/. Accessed .« Back to 2018 ACR/ARHP Annual Meeting
ACR Meeting Abstracts - https://acrabstracts.org/abstract/characterization-of-the-esophageal-microbiome-in-patients-with-systemic-sclerosis-ssc/