Session Information
Session Type: Abstract Session
Session Time: 3:00PM-3:15PM
Background/Purpose: Juvenile idiopathic arthritis (JIA) is the most common chronic pediatric rheumatic disease, yet the spatial immune architecture of inflamed synovium remains poorly characterized. Prior studies in adult rheumatoid arthritis (RA) have revealed spatial niches of immune–stromal interactions that correlate with treatment response. However, whether similar pathogenic circuits exist in JIA is unknown. We aimed to map cellular ecosystems in the JIA joint using high-resolution spatial transcriptomics and to identify microanatomical features associated with inflammation and disease-relevant cell–cell interactions.
Methods: Synovial tissue biopsies were obtained from 9 children with oligoarticular or RF-negative polyarticular JIA (Fig. 1A). Subcellular-resolution spatial transcriptomic profiling was performed using the 10x Genomics Xenium Prime 5K platform, capturing >430,000 cells across all samples. Data were processed using a custom pipeline enabling cell-type annotation, spatial neighborhood enrichment, and ligand–receptor colocalization analysis.
Results: We identified diverse immune and stromal cell populations, including pro-inflammatory macrophages, THY1+NOTCH3+ sublining fibroblasts, and cytotoxic T cells (Fig. 1B-H). Covarying neighborhood analysis revealed sublining fibroblasts and endothelial cells were enriched in JIA synovium with systemic high-inflammatory state (Fig. 1I-J). A newly developed R package, named Spatial Neighborhood Analysis (SNA) (Fig. 2A), revealed cell-cell spatial interaction in JIA synovium (Fig. 2B), including fibroblast–endothelial niches with enriched NOTCH3 signaling, paralleling RA pathology (Fig. 2C-F). Analysis focusing on macrophages showed that pro-inflammatory macrophages were enriched in regions distal from vasculature but proximal to GZMB+CD8+ T cells (Fig. 2G-I), with colocalization scores strongly implicating the CXCL9–CXCR3 axis in macrophage–T cell crosstalk (Fig. 2J-M). The SNA framework also automatically captured tertiary lymphoid structures (TLS) in JIA synovium (Fig. 3A), with dense B and T cell colocalization and CXCL13-CXCR5 signalling (Fig. 3B-H), suggesting active germinal centre-like activity. Moreover, integration with public RA-synovium single-cell data revealed enrichment of NOTCH3+ fibroblasts in JIA, whereas Tph/Tfh and GZMB+ CD8 T cells were more abundant in RA (Fig. 3I-K).
Conclusion: Subcellular spatial transcriptomics uncovers conserved and disease-specific immune–stromal architectures in JIA synovium. Vascular-fibroblast niches and macrophage–T cell crosstalk may be key drivers of inflammation. The presence of TLS and unique myeloid–stromal features in JIA highlight potential biomarkers and therapeutic targets distinct from adult RA. These findings offer new insight into pediatric synovial immunopathology and pave the way for precision therapies in childhood arthritis.
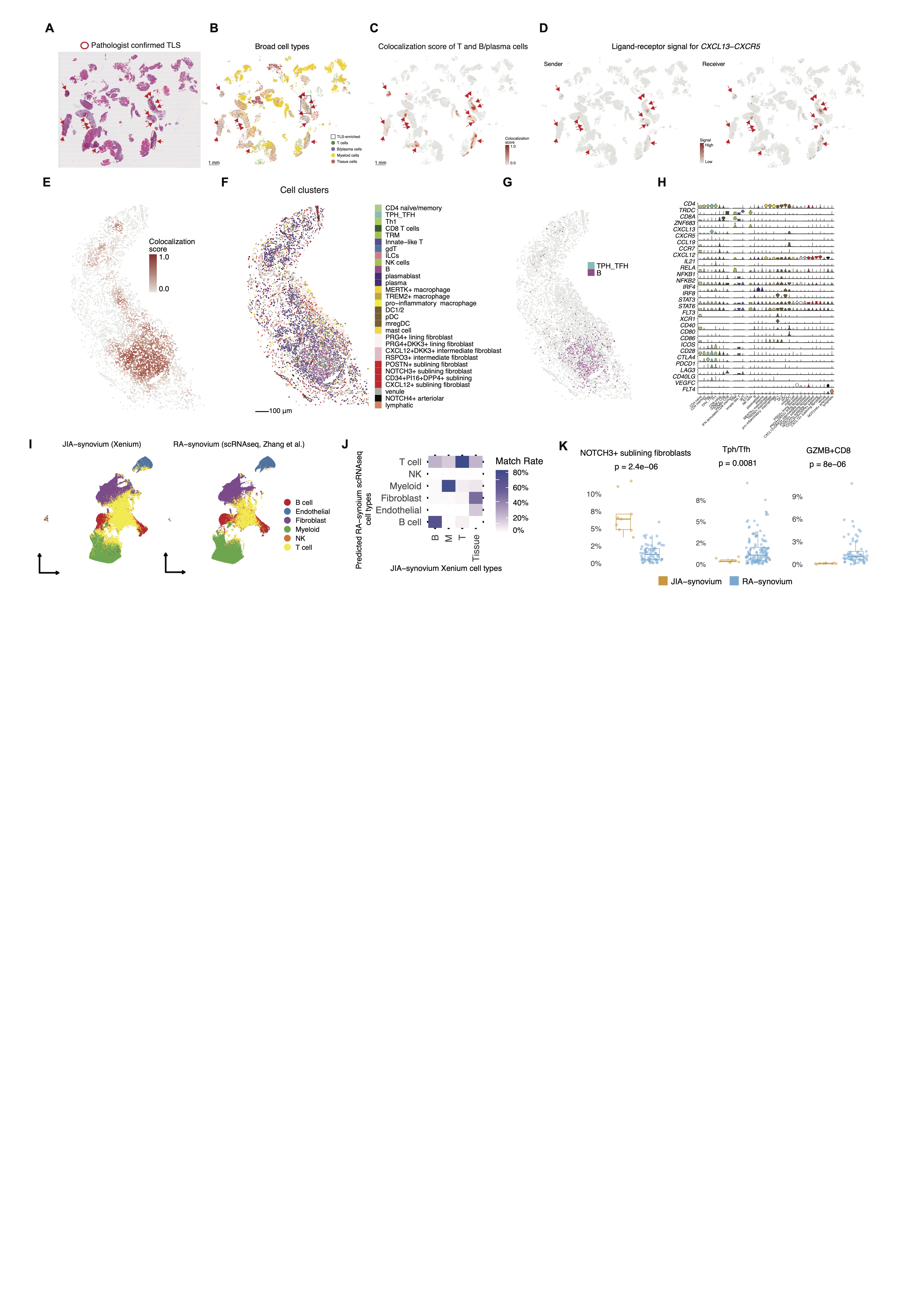 Fig.1: Spatial transcriptomic profiling of synovial tissue in JIA patients. A, Synovial biopsy samples were collected from nine patients diagnosed with JIA. Subcellular resolution spatial transcriptomic data was acquired from FFPE biopsy samples using the 10X Xenium Prime 5K platform. Cell types within synovial tissues were identified, and spatial neighborhoods were characterized based on spatial proximity analysis. B, Identified major immune and tissue-associated cellular compartments in synovium, including T cells, B cells, myeloid cells, and stromal tissue cells (endothelial and fibroblast subsets). Within each broad cell type, fine-scale cell subpopulations were annotated. C, Composition of cell-types across samples. D, Representative example of the FFPE histological slide of synovial biopsy sample and spatial mapping of identified cell clusters. Colors are corresponding with panel B. E-H, Identified fine-scaled cell clusters on UMAP (left) and composition across samples (right) for T cells (E), Myeloid cells (F), B/plasma cells (G), and stromal tissue cells (H). I, Heatmap of covarying neighborhood analysis (CNA) associations of specific cell states with each clinical variable. All testing was adjusted for age and sex. Colours represent the percentage of cell neighbourhoods from each cell state with positive phenotype correlations from white to red (expanded) or blue (depleted). Clinical variables associated globally (permutation p-value < 0.05) are annotated by asterisk (*). J, CNA for CRP association with cell states. Cells in UMAP are colored for expansion (red) or depletion (blue).
Fig.1: Spatial transcriptomic profiling of synovial tissue in JIA patients. A, Synovial biopsy samples were collected from nine patients diagnosed with JIA. Subcellular resolution spatial transcriptomic data was acquired from FFPE biopsy samples using the 10X Xenium Prime 5K platform. Cell types within synovial tissues were identified, and spatial neighborhoods were characterized based on spatial proximity analysis. B, Identified major immune and tissue-associated cellular compartments in synovium, including T cells, B cells, myeloid cells, and stromal tissue cells (endothelial and fibroblast subsets). Within each broad cell type, fine-scale cell subpopulations were annotated. C, Composition of cell-types across samples. D, Representative example of the FFPE histological slide of synovial biopsy sample and spatial mapping of identified cell clusters. Colors are corresponding with panel B. E-H, Identified fine-scaled cell clusters on UMAP (left) and composition across samples (right) for T cells (E), Myeloid cells (F), B/plasma cells (G), and stromal tissue cells (H). I, Heatmap of covarying neighborhood analysis (CNA) associations of specific cell states with each clinical variable. All testing was adjusted for age and sex. Colours represent the percentage of cell neighbourhoods from each cell state with positive phenotype correlations from white to red (expanded) or blue (depleted). Clinical variables associated globally (permutation p-value < 0.05) are annotated by asterisk (*). J, CNA for CRP association with cell states. Cells in UMAP are colored for expansion (red) or depletion (blue).
.jpg) Fig.2: Spatial neighborhood enrichment and niche characterization in JIA synovium. A, Schematic description of the spatial neighborhood enrichment analysis pipeline. The procedure includes identifying nearest neighbors, quantifying neighborhood cell-type composition, performing permutation tests by shuffling cell-type labels, and calculating enrichment Z-scores. B, Spatial neighborhood enrichment in JIA synovium. Statistical significance is indicated as * (adjusted p-value < 0.1 by Benjamini-Hochberg method) and ** (adjusted p-value < 0.05). The highlighted black square indicates the niche of sub-lining fibroblasts and endothelial cells. C, Heatmap displaying the relationship between distances to nearest endothelial cells (purple bar above, 0–300 µm) and fibroblast gene expression. Rows represent fibroblast-associated genes, and columns represent individual cells. Fibroblast marker genes are labeled. Line plots depicting binned distance to nearest endothelial cells (x-axis) and normalized gene expression (y-axis) of representative fibroblast genes. Correlation coefficients and p-values indicate statistical significance. D, Heatmap of gene expressions related to Notch signaling pathway components across different cell types. E, Representative spatial coordinates of a identified niche of sub-lining fibroblasts and endothelial cells, matching pathological images. Cells are colored according to their cluster assignments. F. Spatial expression patterns of fibroblast-related and Notch signaling-related genes within the identified niche location. G, Scaled gene expression of selected M2-like (left) and M1-like (right) macrophage markers plotted as a function of distance from the nearest endothelial cell. Center heatmap shows scaled expression of macrophage-related genes in individual cells ordered by proximity to endothelial cells (0–300 µm), with representative M2- and M1-associated genes annotated. H–I, Correlation between M1-like macrophage module scores and distance to endothelial cells (H) or GZMB+ CD8 T cells (I). Each dot represents a cell. Spearman’s correlation coefficient and p-values are shown. J, Schematic illustrating spatial colocalization scoring between anchor cells and target cells within a 10 µm radius. K, Spatial heatmap of colocalization scores between T cells and macrophages. L, Spatial map of broad cell type annotation including T cells, myeloid cells, B/plasma cells, and tissue cells. M, Spatial ligand–receptor analysis results for CXCL9–CXCR3, which exhibited the strongest correlation with macrophage–T cell co-localization scores.
Fig.2: Spatial neighborhood enrichment and niche characterization in JIA synovium. A, Schematic description of the spatial neighborhood enrichment analysis pipeline. The procedure includes identifying nearest neighbors, quantifying neighborhood cell-type composition, performing permutation tests by shuffling cell-type labels, and calculating enrichment Z-scores. B, Spatial neighborhood enrichment in JIA synovium. Statistical significance is indicated as * (adjusted p-value < 0.1 by Benjamini-Hochberg method) and ** (adjusted p-value < 0.05). The highlighted black square indicates the niche of sub-lining fibroblasts and endothelial cells. C, Heatmap displaying the relationship between distances to nearest endothelial cells (purple bar above, 0–300 µm) and fibroblast gene expression. Rows represent fibroblast-associated genes, and columns represent individual cells. Fibroblast marker genes are labeled. Line plots depicting binned distance to nearest endothelial cells (x-axis) and normalized gene expression (y-axis) of representative fibroblast genes. Correlation coefficients and p-values indicate statistical significance. D, Heatmap of gene expressions related to Notch signaling pathway components across different cell types. E, Representative spatial coordinates of a identified niche of sub-lining fibroblasts and endothelial cells, matching pathological images. Cells are colored according to their cluster assignments. F. Spatial expression patterns of fibroblast-related and Notch signaling-related genes within the identified niche location. G, Scaled gene expression of selected M2-like (left) and M1-like (right) macrophage markers plotted as a function of distance from the nearest endothelial cell. Center heatmap shows scaled expression of macrophage-related genes in individual cells ordered by proximity to endothelial cells (0–300 µm), with representative M2- and M1-associated genes annotated. H–I, Correlation between M1-like macrophage module scores and distance to endothelial cells (H) or GZMB+ CD8 T cells (I). Each dot represents a cell. Spearman’s correlation coefficient and p-values are shown. J, Schematic illustrating spatial colocalization scoring between anchor cells and target cells within a 10 µm radius. K, Spatial heatmap of colocalization scores between T cells and macrophages. L, Spatial map of broad cell type annotation including T cells, myeloid cells, B/plasma cells, and tissue cells. M, Spatial ligand–receptor analysis results for CXCL9–CXCR3, which exhibited the strongest correlation with macrophage–T cell co-localization scores.
.jpg) Fig.3: Identification and characterization of tertiary lymphoid structures (TLS) using T and B cell colocalization scores in JIA synovium and comparative analysis of JIA and RA synovium. A, Images of synovial tissue, illustrating representative TLS as identified by a blinded pathologist. B, Spatial distribution of broad-scale T and B cell colocalization scores. Scale bar, 1 mm. The highlighted black square indicates the TLS-enriched region. C, Colocalization scores for T and B cells. D, Spatial ligand–receptor interactions specifically involving CXCL13-CXCR5. E, Zoomed-in visualization of the area with the highest TLS density highlighted in panel B, colored by T and B cell colocalization score. F-G, Same region as in panel E, displayed according to fine-scale cell cluster assignments. H, Violin plots showing the expression levels of key TLS-related genes across different cell clusters identified within the TLS. I, Cell-type level label transfer results, where RA synovium scRNA-seq (Zhang et al.) was used as the reference and JIA synovium Xenium data as the query. RA-derived annotations were transferred to individual JIA cells using a k-nearest neighbors (kNN) approach in the integrated latent space. J, Match rate between transferred RA scRNA-seq cell types and original Xenium-based annotations in JIA synovium. Matrix shows percentage overlap between each transferred and original broad cell type. K, Box plots showing the proportion of cell clusters across individual samples, stratified by condition (JIA vs RA synovium). P-values were calculated using Wilcoxon rank-sum tests.
Fig.3: Identification and characterization of tertiary lymphoid structures (TLS) using T and B cell colocalization scores in JIA synovium and comparative analysis of JIA and RA synovium. A, Images of synovial tissue, illustrating representative TLS as identified by a blinded pathologist. B, Spatial distribution of broad-scale T and B cell colocalization scores. Scale bar, 1 mm. The highlighted black square indicates the TLS-enriched region. C, Colocalization scores for T and B cells. D, Spatial ligand–receptor interactions specifically involving CXCL13-CXCR5. E, Zoomed-in visualization of the area with the highest TLS density highlighted in panel B, colored by T and B cell colocalization score. F-G, Same region as in panel E, displayed according to fine-scale cell cluster assignments. H, Violin plots showing the expression levels of key TLS-related genes across different cell clusters identified within the TLS. I, Cell-type level label transfer results, where RA synovium scRNA-seq (Zhang et al.) was used as the reference and JIA synovium Xenium data as the query. RA-derived annotations were transferred to individual JIA cells using a k-nearest neighbors (kNN) approach in the integrated latent space. J, Match rate between transferred RA scRNA-seq cell types and original Xenium-based annotations in JIA synovium. Matrix shows percentage overlap between each transferred and original broad cell type. K, Box plots showing the proportion of cell clusters across individual samples, stratified by condition (JIA vs RA synovium). P-values were calculated using Wilcoxon rank-sum tests.
To cite this abstract in AMA style:
Inamo J, Fierkens R, Clay M, Lin C, Hayes K, Rogers N, Leach H, Yomogida K. Subcellular resolution spatial transcriptomics reveals immune-stromal crosstalk within the synovium of patients with juvenile idiopathic arthritis [abstract]. Arthritis Rheumatol. 2025; 77 (suppl 9). https://acrabstracts.org/abstract/subcellular-resolution-spatial-transcriptomics-reveals-immune-stromal-crosstalk-within-the-synovium-of-patients-with-juvenile-idiopathic-arthritis/. Accessed .« Back to ACR Convergence 2025
ACR Meeting Abstracts - https://acrabstracts.org/abstract/subcellular-resolution-spatial-transcriptomics-reveals-immune-stromal-crosstalk-within-the-synovium-of-patients-with-juvenile-idiopathic-arthritis/