Session Information
Session Type: Poster Session B
Session Time: 9:00AM-11:00AM
Background/Purpose: Systemic lupus erythematosus (SLE) is a chronic, systemic autoimmune disorder. The pathogenesis of SLE is not fully understood, but high twin/sibling concordance rates suggest a genetic component triggered by environmental events. Childhood-onset SLE (cSLE) patients have an extreme phenotype, therefore is an ideal population to study genetic contributors to SLE. Historically, studies have focused on unrelated individulas who share variants in individual genes. However, this approach doesn’t consider that rare variants in SLE could cluster in genes participating in related biological processes. We performed genome sequencing in a diverse cohort of cSLE patients and parental controls, and describe a network-of-pathways approach to identify biological pathways enriched in genes with rare variants that may contribute to cSLE pathogenesis.
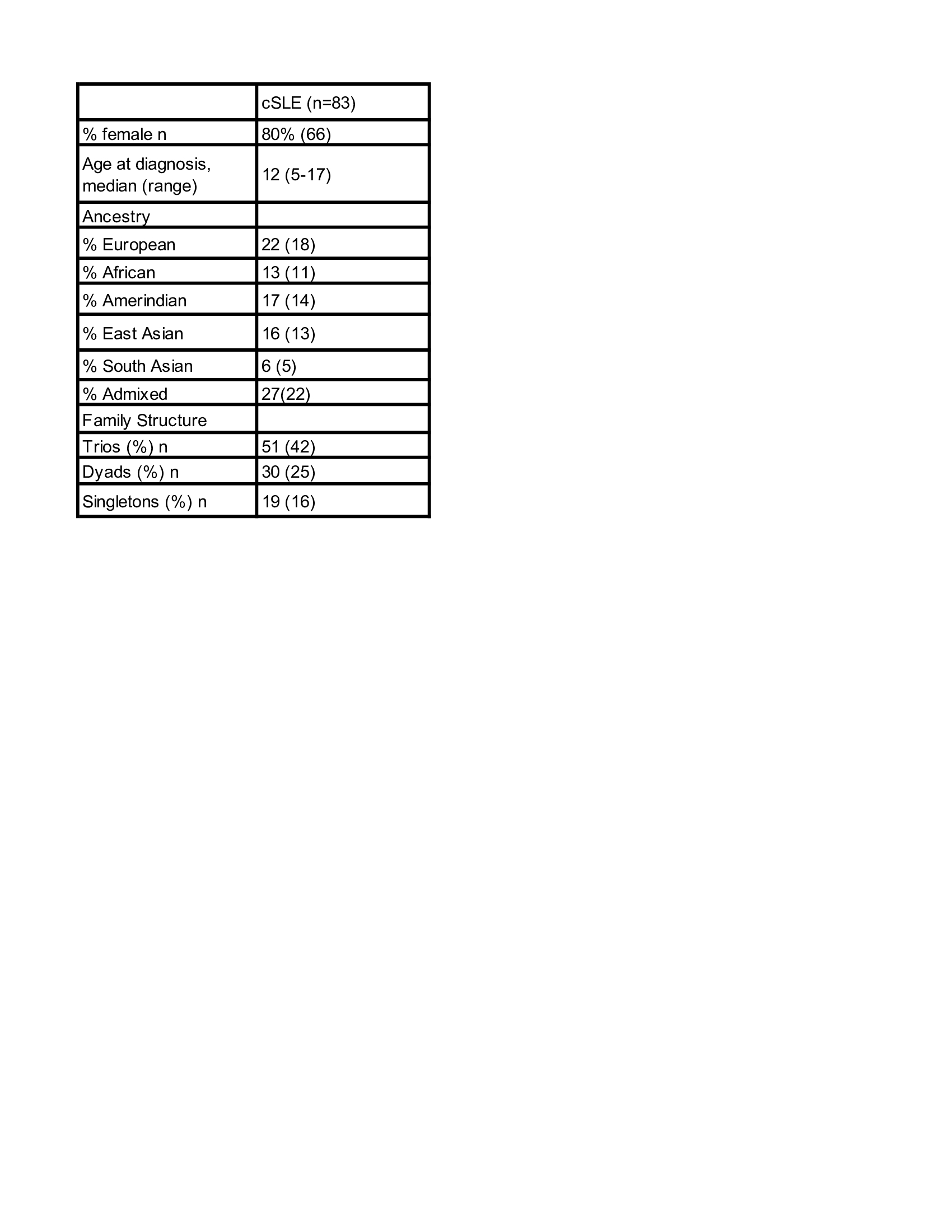
Methods: SLE patients met at least 4 of 11 revised American College of Rheumatology classification criteria, with disease onset 18 years. Whole blood samples were collected from 83 patients with cSLE and 109 unaffected parents and whole genome sequencing was performed. The genome sequences were filtered to select for rare (minor allele frequency< 0.01), nonsynonymous variants in coding exons. Selected variants had to be present only in cSLE patients or in both patients and parents, but at a significantly higher level in patients (Fisher’s exact test (p0.05)).This resulted in 501 unique variants, which corresponded to 232 genes.Using the 232 genes, we performed pathway enrichment analysis based on Gene Ontology biological processes with Metascape. We generate a network of enriched pathways, weighted by enrichment values and overlap coefficients with Cytoscape. Pathway clusters in the network were manually reviewed and classified. We analyzed clinical phenotypes and CADD-Phred for each pathway cluster.
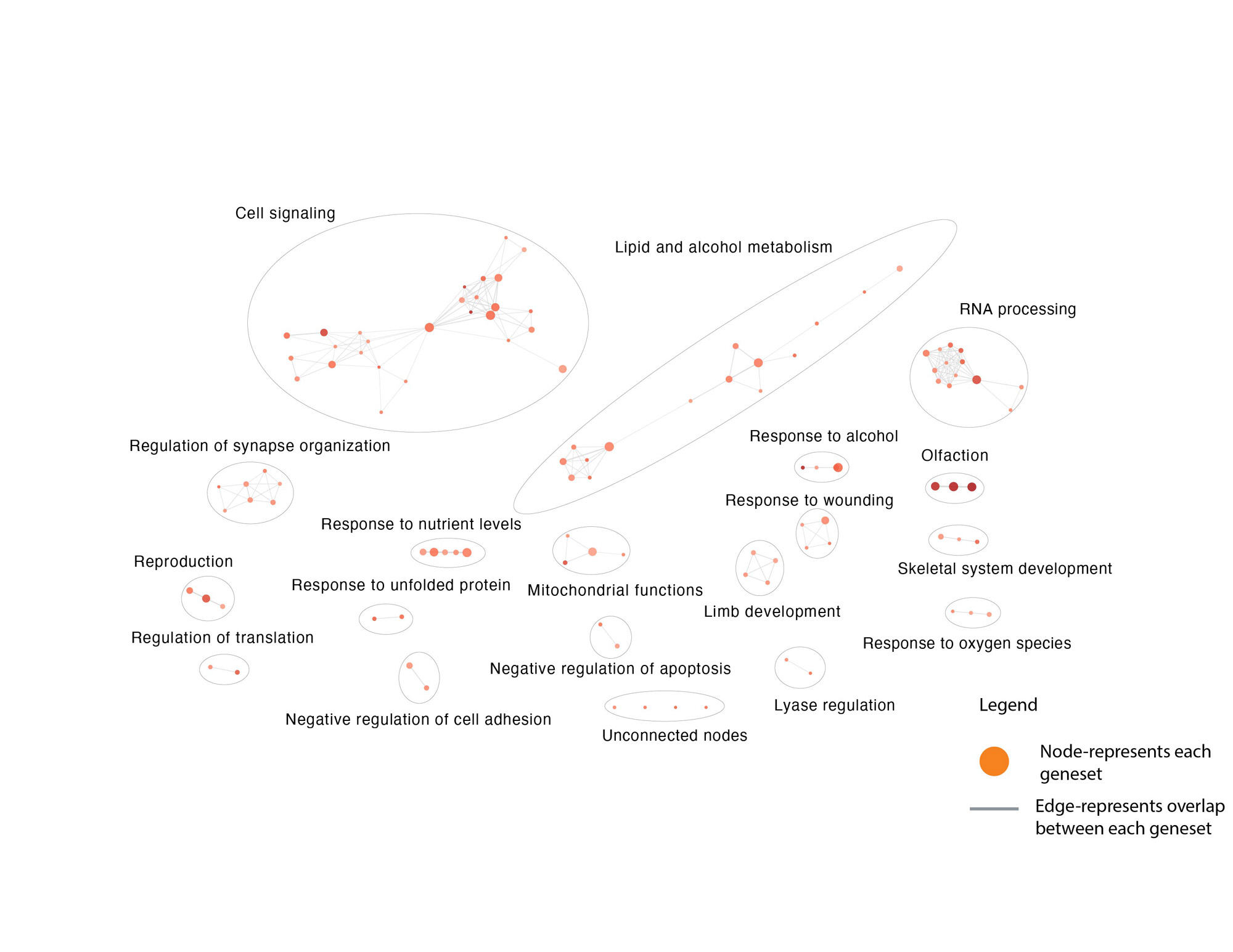
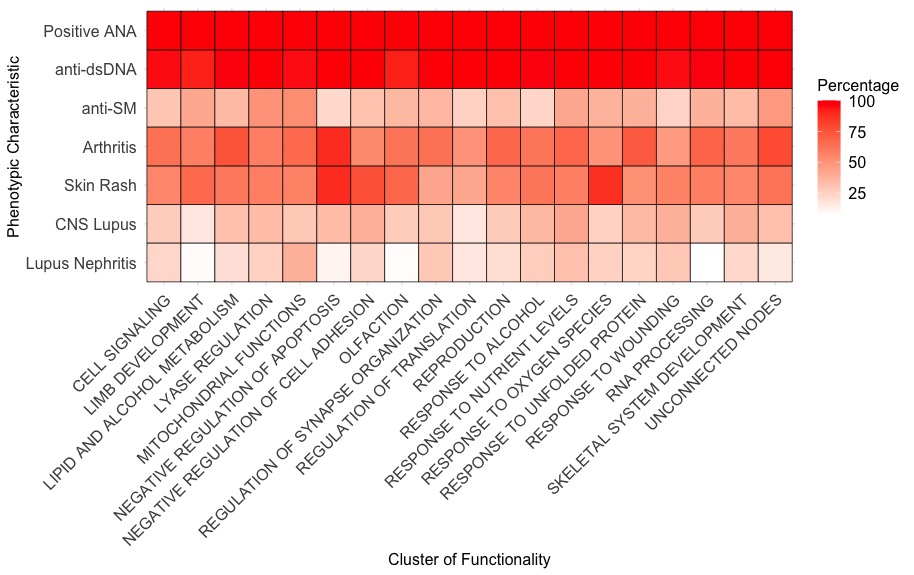
Results: Rare genetic variants in cSLE patients are enriched in biological pathways such as RNA processing and apoptosis (Figure 1). Some pathways(i.e. nucleic acid regulation) are well-established causes of monogenic SLE, while others are novel. The median CADD-Phred score for each of the 18 networks ranges from 11.5-23.6.The mitochondrial functions cluster had higher rates of lupus nephritis, and the cell adhesion cluster had higher rates of CNS SLE compared to other clusters (Figure 2).
Conclusion: Network analysis is a useful approach to identify biological pathways and specific genes that could contribute to cSLE risk. Ongoing detailed analysis of the specific variants identified in each pathway will allow us to prioritize key genes and pathways for further study. In total, this analysis may contribute to advancing the understanding of cSLE beyond a broad clinical phenotype and towards a more precise molecular diagnosis.
To cite this abstract in AMA style:
Heitzman K, Dass S, Hiraki L, Silverman E, Scott C, Barrera-Vargas A, Deng Z, Kaplan M, Franco L, Lewandowski L. Network Analysis of Genome Sequences Identifies Important Pathways in the Pathogenesis of Childhood-onset Systemic Lupus Erythematosus [abstract]. Arthritis Rheumatol. 2023; 75 (suppl 9). https://acrabstracts.org/abstract/network-analysis-of-genome-sequences-identifies-important-pathways-in-the-pathogenesis-of-childhood-onset-systemic-lupus-erythematosus/. Accessed .« Back to ACR Convergence 2023
ACR Meeting Abstracts - https://acrabstracts.org/abstract/network-analysis-of-genome-sequences-identifies-important-pathways-in-the-pathogenesis-of-childhood-onset-systemic-lupus-erythematosus/