Session Information
Date: Sunday, November 10, 2019
Title: Pediatric Rheumatology – ePoster I: Basic Science, Biomarkers, & Sclerodermic Fever
Session Type: Poster Session (Sunday)
Session Time: 9:00AM-11:00AM
Background/Purpose: Hypocomplementemic urticarial vasculitis syndrome (HUVS) is a rare disease characterized by persistent urticarial lesions and hypocomplementemia associated with systemic features involving musculoskeletal, pulmonary, renal and gastrointestinal systems. Systemic lupus erythematosus (SLE) develops in >50% of patients with HUVS, although the pathogenesis is unknown.
Methods: We describe 6 paediatric patients with HUVS, three of whom carry a homozygous variant of DNASE1L3 and present a peculiar clinical phenotype. A Targeted Resequencing using a panel including genes already known to be mainly associated to Interferonopathies Lupus-like (DNASE1, DNASE2, DNASE1L3, TREX1) on the Illumina NextSeq® platform was performed. All variants identified were confirmed by Sanger sequencing and, when possible, family members were tested to study the segregation of identified variants. We applied in silico studies only to variants with an allelic frequency ≤1%.
Results: All patients described are Caucasian and 3 of them are female. Two patients presented at onset with extended cutaneous manifestation, joints and abdominal involvement with cholecystitis. They did not develop renal or pulmonary involvement. In contrast, the other four patients presented a more severe disease. All of them developed renal involvement (from microhaematuria up to nephrotic syndrome) with renal biopsy showing mesangial glomerulonephritis in three patients and pauci-immune glomerulonephritis (ANCA negative) in one. Moreover, two of them developed also pulmonary vasculitis (Table 1). A homozygous DNASE1L3 variant (c.290_291delCA) was identified in three of these patients. All of them were treated with glucocorticoid and dapsone at onset. Cyclophosphamide, mycophenolate mofetil and azathioprine were used in patients with renal involvement. None of them developed SLE.
Conclusion: HUVS is very rare disease in childhood. Approximately 50% of HUVS patients develop SLE. Genetic susceptibility to SLE is recognized and DNASE1L3-related SLE have been reported. Özçakar et al. have described 5 children from two families with HUVS who carry the same variant on DNASE1L3 that we report here (1). Our patients confirm that variant in DNASE1L3 can cause HUVS and support the hypothesis that this variant is responsible of a more severe phenotype with major organ involvement (renal and pulmonary). Patients with HUVS need to be followed very strictly for the risk to develop SLE. Presence of variant in DNASE1L3 can identify patients with more severe disease and high risk to develop major organ involvement. These patients need more aggressive and possibly life-long immunosuppressive treatment.
Reference.
(1) Ozçakar ZB et al. DNASE1L3 Mutations in Hypocomplementemic Urticarial Vasculitis Syndrome. Arthritis Rheum. 2013 Aug;65(8):2183-9.
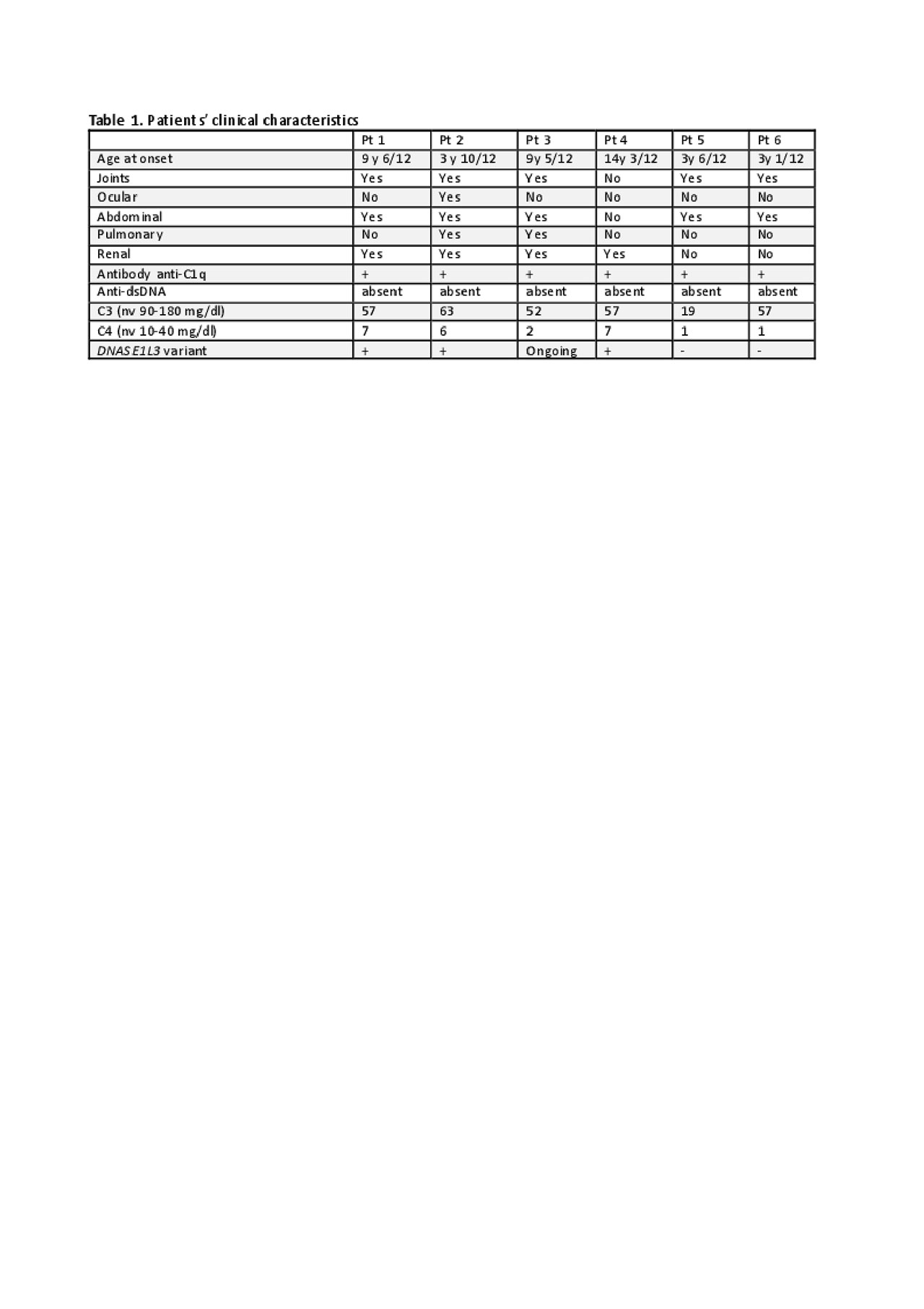
TABLE1_HUVS_IMMAGE
To cite this abstract in AMA style:
Ranalli M, Passarelli C, Messia V, Pardeo M, Sacco E, Insalaco A, Vivarelli M, De Benedetti F, Bracaglia C. DNASE1L3 Variant in Hypocomplementemic Urticarial Vasculitis Syndrome Identifies a Different Clinical Phenotype [abstract]. Arthritis Rheumatol. 2019; 71 (suppl 10). https://acrabstracts.org/abstract/dnase1l3-variant-in-hypocomplementemic-urticarial-vasculitis-syndrome-identifies-a-different-clinical-phenotype/. Accessed .« Back to 2019 ACR/ARP Annual Meeting
ACR Meeting Abstracts - https://acrabstracts.org/abstract/dnase1l3-variant-in-hypocomplementemic-urticarial-vasculitis-syndrome-identifies-a-different-clinical-phenotype/