Session Information
Date: Tuesday, November 10, 2015
Title: Systemic Sclerosis, Fibrosing Syndromes and Raynaud's - Clinical Aspects and Therapeutics Poster III
Session Type: ACR Poster Session C
Session Time: 9:00AM-11:00AM
Background/Purpose:
Pulmonary arterial hypertension (PAH) is an important
complication in patients with connective tissue disease and may lead to
increased morbidity and early mortality. Ambrisentan, an orally active
endothelin A receptor selective antagonist, has demonstrated safety and
efficacy in patients with WHO Group I PAH. Due to the chronic, long-term nature
of treatment, dose adjustments are often desired to enhance efficacy or
attenuate adverse events. We analyzed the ARIES database to determine dose
adjustments in patients with CTD-PAH treated with ambrisentan over a three year
period.
Methods:
Data from the combined ARIES 1 & 2 placebo-controlled
studies (ARIES-C) as well as the long-term extension study (ARIES-E) were
evaluated. Available ambrisentan dosages in ARIES-C were 2.5mg, 5mg, and 10mg
once daily. Patients randomized to placebo in ARIES-C who continued to ARIES-E
were re-randomized to active treatment. ARIES-E was open-label; however
subjects and investigators were blinded to dose for the first 24 weeks of
treatment during which a single, blinded dose reduction was permitted in the
case of study drug intolerance. After 24 weeks, dosage adjustments were
permitted at the discretion of the investigator. The dosage of ambrisentan was
recorded at each study visit for all patients continuing in the study and on
therapy. At the end of 3 years, survival status was collected retrospectively
for patients no longer participating in the study.
Results:
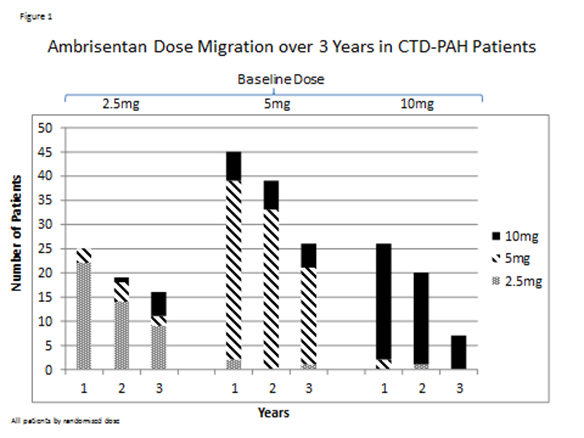
There were 124 patients with CTD-PAH who received at least
one dose of ambrisentan. The numbers of patients randomized to receive each
initial dose were: 2.5mg = 30, 5mg = 60, and 10mg = 34. Annual patient
disposition is outlined in Table 1. Over the course of three years,
approximately 40% (n=49) of patients continued to receive ambrisentan therapy
in ARIES-E. Reasons for discontinuation included transitioning out of the
study (n=28, 23%) or terminating participation (n=45, 36%). Retrospectively collected
survival status at 3 years revealed 83 patients (67%) were still alive, 29
patients (23%) had died, and survival status was unknown in 12 patients (10%). Dose
adjustments of ambrisentan over the course of three years are represented in
Figure 1.
Conclusion:
Through three years of treatment with ambrisentan, those
patients with CTD-PAH remaining in ARIES-E predominately remained on the
dosages they were originally randomized to.
Table 1
|
|
Year 1 |
Year 2 |
Year 3 |
|||
|
|
Ongoing |
Not Ongoing* |
Ongoing |
Not Ongoing* |
Ongoing |
Not Ongoing* |
|
2.5mg (n = 30) |
25 |
5 |
19 |
11 |
16 |
14 |
|
5mg (n = 60) |
45 |
15 |
39 |
21 |
26 |
34 |
|
10mg (n = 34) |
26 |
8 |
20 |
14 |
7 |
27 |
*Values of patients not ongoing for each dosage are
cumulative
To cite this abstract in AMA style:
Tislow J, Blair C, Gillies H. Ambrisentan Dose Migration over 3 Years in Patients with Connective Tissue Disease-Associated Pulmonary Arterial Hypertension (CTD-PAH) [abstract]. Arthritis Rheumatol. 2015; 67 (suppl 10). https://acrabstracts.org/abstract/ambrisentan-dose-migration-over-3-years-in-patients-with-connective-tissue-disease-associated-pulmonary-arterial-hypertension-ctd-pah/. Accessed .« Back to 2015 ACR/ARHP Annual Meeting
ACR Meeting Abstracts - https://acrabstracts.org/abstract/ambrisentan-dose-migration-over-3-years-in-patients-with-connective-tissue-disease-associated-pulmonary-arterial-hypertension-ctd-pah/