Session Information
Session Type: Poster Session D
Session Time: 8:30AM-10:30AM
Background/Purpose: Alteration of the gut microbiome has been linked to the pathogenesis of systemic lupus erythematosus (SLE). However, a comprehensive view of the gut microbiome in SLE and its interaction with the host remains to be revealed. This study aimed to reveal SLE-associated changes in the gut microbiome and its interaction with the host by a comprehensive metagenome-wide association study (MWAS) based on shotgun sequencing followed by integrative analysis.
Methods: We performed a MWAS of SLE based on shotgun sequencing of the gut microbial DNA from Japanese individuals (Ncase = 47, Ncontrol = 203). All of the 47 SLE patients were diagnosed according to the systemic lupus international collaborating clinics classification criteria (SLICC). Our MWAS consisted of three major bioinformatic analytic pipelines (phylogenetic analysis, functional gene analysis and pathway analysis). We integrated the result of the MWAS with the genome-wide association study (GWAS) data and plasma metabolite data.
Results: Via species level phylogenetic analysis, we identified and validated increases of Streptococcus interdemius and Streptococcus anginosus in the SLE patients (effect size = 0.617 and Pmicrobe = 3.7 × 10-5 for Streptococcus anginosus, effect size = 0.579 and Pmicrobe = 7.5 × 10-5 for Streptococcus intermedius; Figure 1). Microbial gene analysis revealed increases of the eight Streptococcus derived genes including one involved in redox reaction. Additionally, microbial pathways related to sulfur metabolism and flagella assembly were altered in the SLE patients. We identified a pathway level SLE-specific link between the metagenome and the germline genome by comparing the result of the current MWAS and the GWAS of SLE (i.e., MWAS-GWAS interaction). α- and β-diversity analyses provided evidence of dysbiosis in the metagenome of the SLE patients (Figure 2). Microbiome-metabolome association analysis identified positive dosage correlation of acylcarnitine with Streptococcus intermedius, an SLE-associated taxon (Figure 3).
Conclusion: Our MWAS followed by integrative analysis revealed SLE-associated changes in the gut microbiome and its interaction with the host, which provided novel insights into the pathogenesis of SLE.
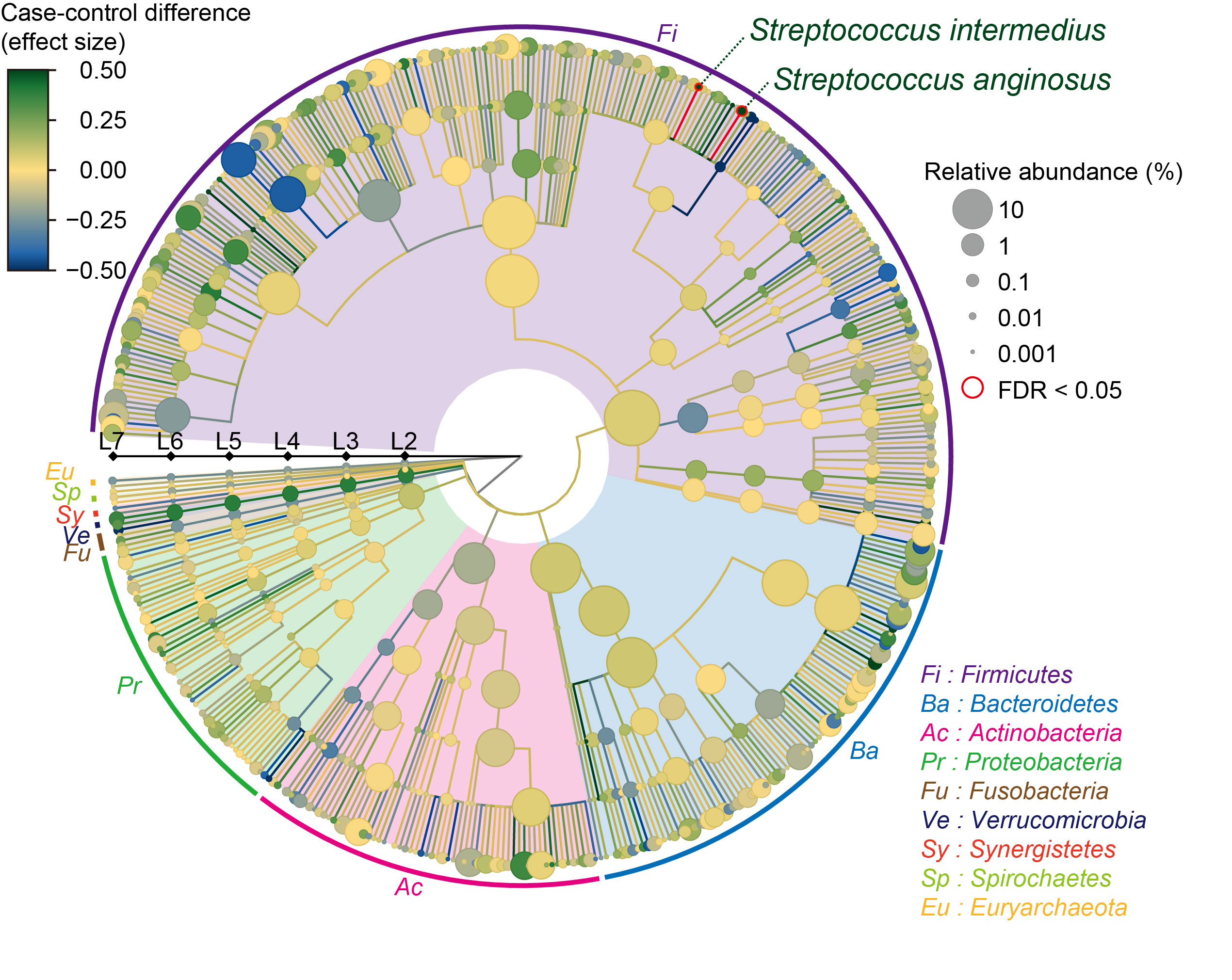 A phylogenetic tree. Levels L2-L7 are from the inside layer to the outside layer. The size and the color of nodes represent relative abundances and effect sizes, respectively. The two clades with significant case-control associations (FDR < 0.05) are outlined in red. FDR, false discovery ratio.
A phylogenetic tree. Levels L2-L7 are from the inside layer to the outside layer. The size and the color of nodes represent relative abundances and effect sizes, respectively. The two clades with significant case-control associations (FDR < 0.05) are outlined in red. FDR, false discovery ratio.
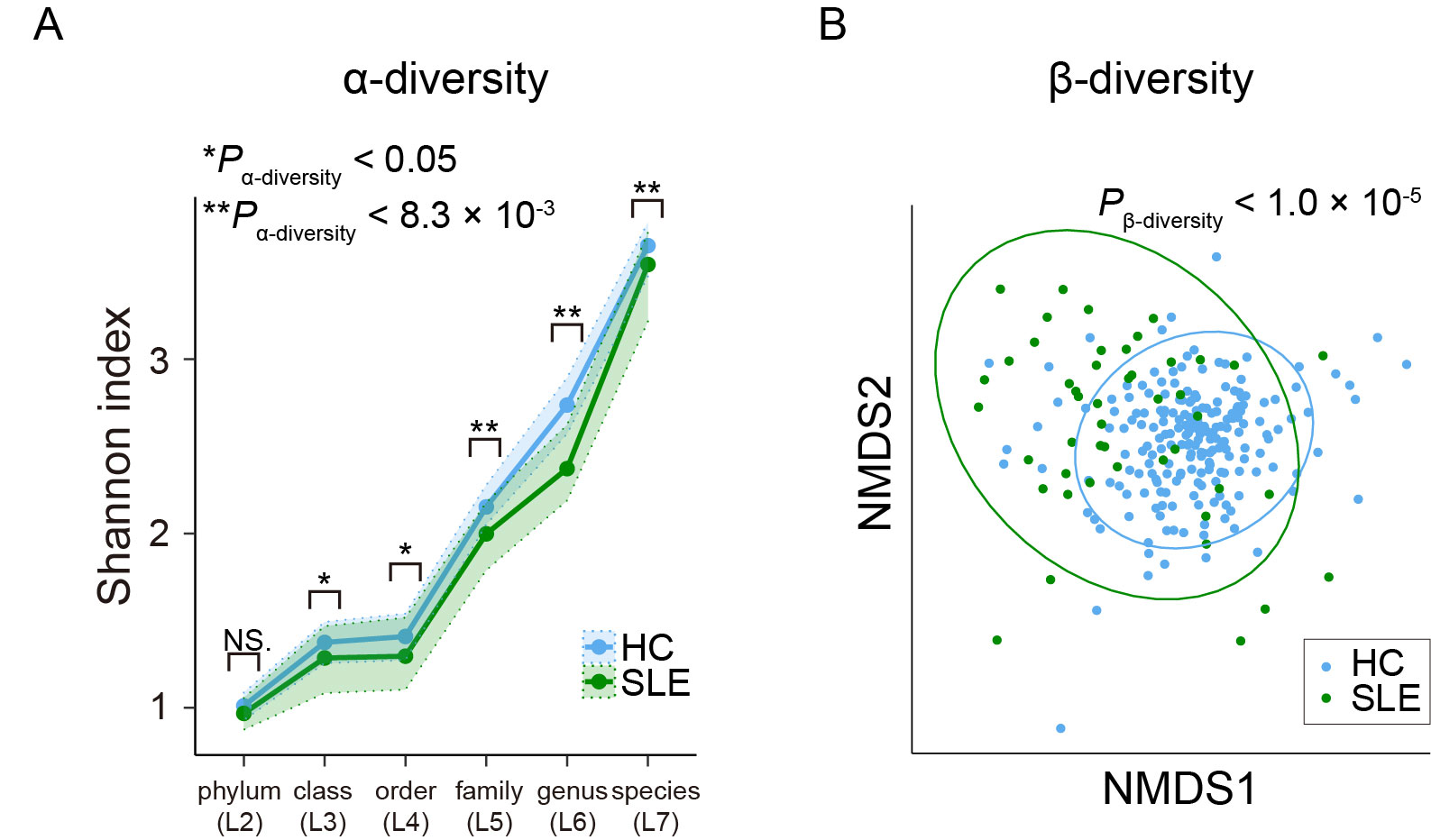 (A) α-diversities of the phylogenetic relative abundance data for the six taxonomic levels. Blue and green dots represent the median Shannon index of the HC and SLE subjects. Upper and lower dashed lines indicate the 1st and 3rd quantile of Shannon index for the HC and SLE subjects. (B) β-diversities of the phylogenetic relative abundance data at the species level. Result of NMDS based on Bray-Curtis distance is represented. Blue and green dots represent the HC and SLE subjects. HC, healthy control; IQR, interquartile range; NMDS, non-metric multidimensional scaling; SLE, systemic lupus erythematosus; *, P < 0.05; **, P < 0.0083.
(A) α-diversities of the phylogenetic relative abundance data for the six taxonomic levels. Blue and green dots represent the median Shannon index of the HC and SLE subjects. Upper and lower dashed lines indicate the 1st and 3rd quantile of Shannon index for the HC and SLE subjects. (B) β-diversities of the phylogenetic relative abundance data at the species level. Result of NMDS based on Bray-Curtis distance is represented. Blue and green dots represent the HC and SLE subjects. HC, healthy control; IQR, interquartile range; NMDS, non-metric multidimensional scaling; SLE, systemic lupus erythematosus; *, P < 0.05; **, P < 0.0083.
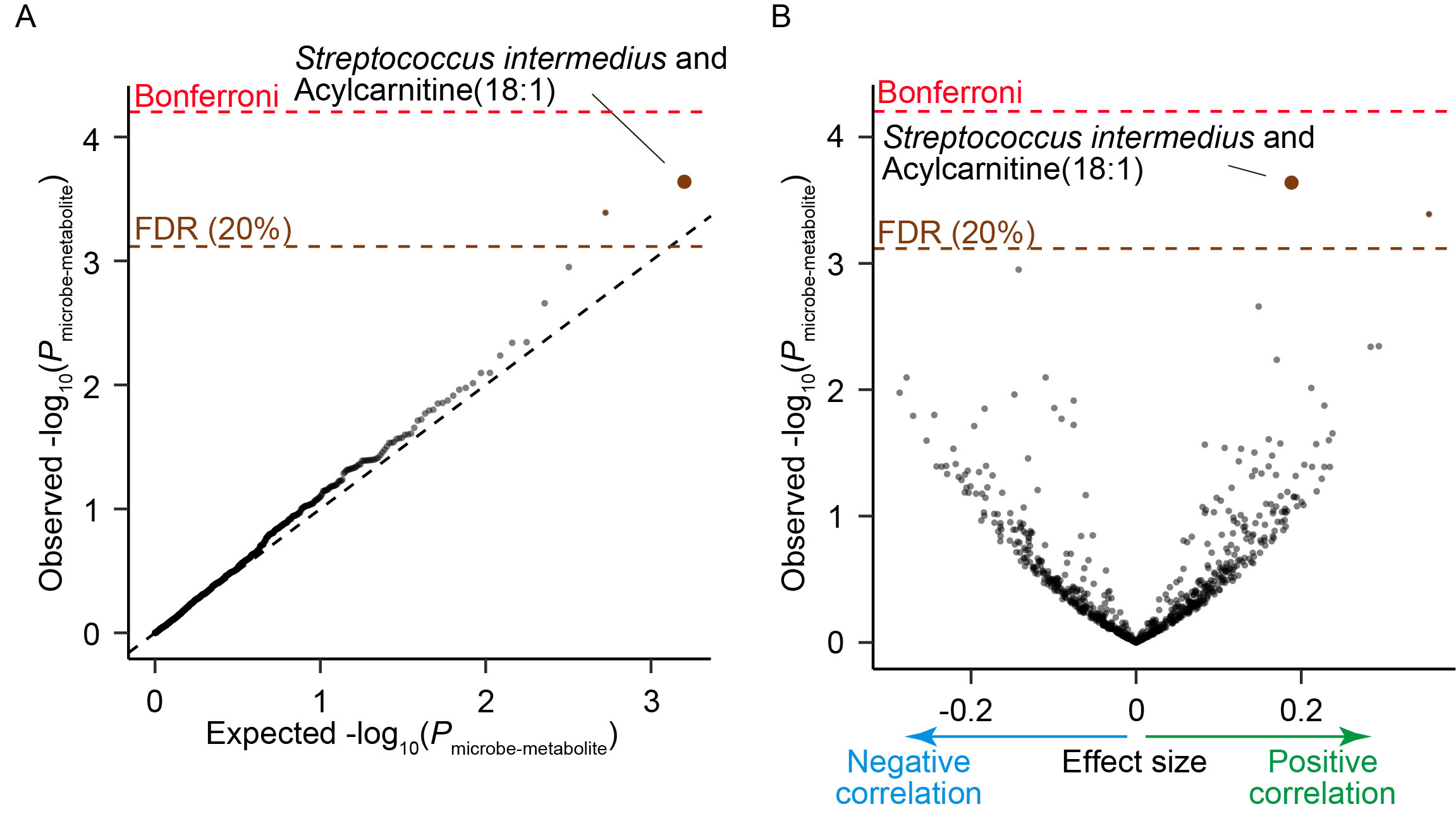 (A) A quantile-quantile plot of the p-values from the microbe-metabolite association analysis. The x-axis indicates log-transformed empirically estimated median P-value. The y-axis indicates observed -log10(P). The diagonal dashed line represents y = x, which corresponds to the null hypothesis. The horizontal red dashed line indicates the Bonferroni-corrected threshold (α = 0.05), and the brown dashed line indicates the FDR threshold (FDR = 0.20) calculated with Benjamini-Hochberg method. The microbe-metabolite pairs with FDR < 0.20 are plotted as brown dots, and the other microbe-metabolite pairs are plotted as black dots. (B) A volcano plot. The x-axis indicates effect sizes in linear regression. The y-axis, horizontal dashed lines and dot colors are the same as in (A). FDR, false discovery rate; HC, healthy control; SLE, systemic lupus erythematosus.
(A) A quantile-quantile plot of the p-values from the microbe-metabolite association analysis. The x-axis indicates log-transformed empirically estimated median P-value. The y-axis indicates observed -log10(P). The diagonal dashed line represents y = x, which corresponds to the null hypothesis. The horizontal red dashed line indicates the Bonferroni-corrected threshold (α = 0.05), and the brown dashed line indicates the FDR threshold (FDR = 0.20) calculated with Benjamini-Hochberg method. The microbe-metabolite pairs with FDR < 0.20 are plotted as brown dots, and the other microbe-metabolite pairs are plotted as black dots. (B) A volcano plot. The x-axis indicates effect sizes in linear regression. The y-axis, horizontal dashed lines and dot colors are the same as in (A). FDR, false discovery rate; HC, healthy control; SLE, systemic lupus erythematosus.
To cite this abstract in AMA style:
Tomofuji Y, Maeda Y, Oguro-Igashira E, Kishikawa T, Yamamoto K, Sonehara K, Motooka D, Matsumoto Y, Matsuoka H, Yoshimura M, Yagita M, Nii T, Ohshima S, Nakamura S, Inohara H, Takeda K, Kumanogoh A, Okada Y. A Metagenome-wide Association Study Revealed Disease-specific Landscape of the Gut Microbiome of Systemic Lupus Erythematosus in Japanese [abstract]. Arthritis Rheumatol. 2021; 73 (suppl 9). https://acrabstracts.org/abstract/a-metagenome-wide-association-study-revealed-disease-specific-landscape-of-the-gut-microbiome-of-systemic-lupus-erythematosus-in-japanese/. Accessed .« Back to ACR Convergence 2021
ACR Meeting Abstracts - https://acrabstracts.org/abstract/a-metagenome-wide-association-study-revealed-disease-specific-landscape-of-the-gut-microbiome-of-systemic-lupus-erythematosus-in-japanese/