Session Information
Session Time: 6:00PM-7:00PM
Background/Purpose: Insufficiency of Suppressor of Cytokine Signaling 1 (SOCS1) typically manifests with autoimmune features. Recently, some cases have also shown autoinflammatory traits such as recurrent oral stomatitis. However, the functional immunogenetic basis of these autoinflammatory presentations remains poorly defined. We describe a family with SOCS1 haploinsufficiency and provide functional evidence supporting an expanded phenotype that includes autoinflammatory manifestations, from which targeted therapy was derived.
Methods: The proband, a 47-year-old male, presented with severe oral stomatitis and gastric ulcers, alongside leukopenia, type 1 diabetes, and Hashimoto thyroiditis. Three of his four children exhibited overlapping clinical features:
Daughter (age 16): recurrent oral stomatitis, recurrent fevers, ANA positivity, chronic polyarticular arthritis, Hashimoto thyroiditis, and prior Henoch–Schönlein purpura.
Son (age 14) and daughter (age 11): chronic recurrent oral stomatitis.
Trio whole-exome sequencing (WES) was performed on the proband and two affected children. Functional assessment of interferon suppression was performed by transfection of plasmids encoding wildtype or mutant SOCS1 into a reporter cell line. Plasma cytokine profiling was performed using Olink proximity extension assay.
Results: Genetic analysis identified a novel SOCS1 in-frame deletion, c.386_400del (p.His129_Gly133del), affecting a highly conserved region within the SH2 domain. The mutant SOCS1 protein displayed impaired ability to suppress type I, type II and type II IFN signaling (Figure 1). Correspondingly, plasma cytokine profiling revealed elevated levels of IFN-inducible chemokines including CXCL9 and CXCL10 in the affected individuals. In the 16-year-old daughter who received treatment with the JAK inhibitor tofacitinib, clinical improvement correlated with a reduction in these chemokine levels.
Conclusion: This study broadens the phenotypic spectrum of SOCS1 insufficiency to include predominant autoinflammatory manifestations, notably severe oral and upper gastrointestinal ulcerations. Functional characterization of a novel 4–amino acid deletion supports its pathogenicity. Targeted JAK inhibition led to clinical and biochemical improvement, highlighting its therapeutic potential. Future studies involving STAT1 phosphorylation assays and transcriptomic profiling will further elucidate the molecular consequences of this SOCS1 variant and refine treatment approaches for affected individuals.
Figure 1. Mutant SOCS1 showed impaired ability to suppress IFN stimulation. 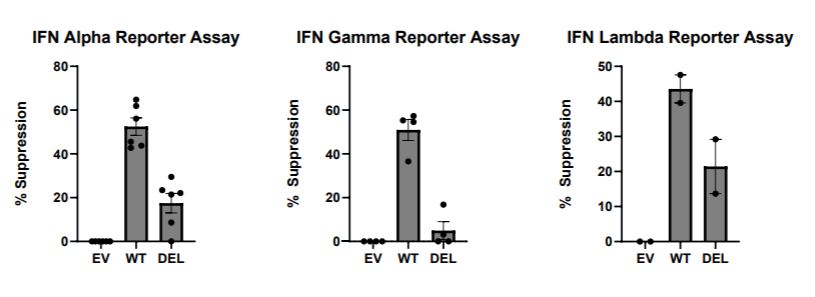 Plasmids encoding wildtype (WT) SOCS1, mutant SOCS1 (DEL; p.His129_Gly133del), or empty vector (EV) were transfected into HEK cell lines with luciferase reporters for IFN alpha, IFN gamma or IFN lambda. Percentage suppression was calculated based on the reduction of luciferase signal compared to EV.
Plasmids encoding wildtype (WT) SOCS1, mutant SOCS1 (DEL; p.His129_Gly133del), or empty vector (EV) were transfected into HEK cell lines with luciferase reporters for IFN alpha, IFN gamma or IFN lambda. Percentage suppression was calculated based on the reduction of luciferase signal compared to EV.
To cite this abstract in AMA style:
Zeft A, Lee P, Vondenberg J, Rimland C, Weng R, Lesmana H. SOCS1 Haploinsufficiency: Functional Evidence Supporting an Expanded Clinical Phenotype [abstract]. Arthritis Rheumatol. 2026; 78 (suppl 3). https://acrabstracts.org/abstract/socs1-haploinsufficiency-functional-evidence-supporting-an-expanded-clinical-phenotype/. Accessed .« Back to 2026 Pediatric Rheumatology Symposium
ACR Meeting Abstracts - https://acrabstracts.org/abstract/socs1-haploinsufficiency-functional-evidence-supporting-an-expanded-clinical-phenotype/